Research Articles
Advancing Drug Discovery: How MC-PDFT Computationally Models Strong Electron Correlation in Biomolecular Systems
This article provides a comprehensive guide for researchers and drug development professionals on the application of Multiconfiguration Pair-Density Functional Theory (MC-PDFT) to strongly correlated electron systems.
Unlocking Diradical Systems: MC-PDFT Accuracy for Drug Discovery and Advanced Materials
This article provides a comprehensive analysis of Multiconfiguration Pair-Density Functional Theory (MC-PDFT) for modeling challenging diradical systems.
MALA: How Materials Learning Algorithms Are Revolutionizing DFT Calculations for Drug Discovery
This article provides a comprehensive overview of Materials Learning Algorithms (MALA) for accelerating Density Functional Theory (DFT) calculations, targeted at computational researchers and drug development professionals.
Level Shifting vs. Damping: A Practical Guide to Mastering SCF Convergence in Computational Chemistry
This article provides a comprehensive, practical guide for computational chemists and materials scientists on two critical techniques for achieving Self-Consistent Field (SCF) convergence: level shifting and damping.
Unlocking Enhanced Sampling in AIMD: The Lagrange Interpolation Molecular Orbital (LIMO) Method Explained
This article provides a comprehensive guide to the Lagrange Interpolation Molecular Orbital (LIMO) method for accelerated ab initio molecular dynamics (AIMD).
LSMO vs LIMO in AIMD Simulations: A Comparative Guide for Biomolecular Dynamics and Drug Discovery
This article provides a comprehensive comparison of the Locally-Sampled Molecular Orbital (LSMO) and Linear-scaling Self-consistent Field with Maximally Localized Molecular Orbitals (LIMO) methods for Ab Initio Molecular Dynamics (AIMD) simulations,...
Accelerating Drug Discovery: Mastering LSMO Methods for Robust SCF Convergence in Biomolecular Geometry Optimization
This article provides a comprehensive guide to the Linear-Scaling Self-Consistent Field (LS-SCF or LSMO) method for achieving reliable Self-Consistent Field (SCF) convergence during geometry optimization, a critical bottleneck in computational...
Integrating LAMMPS with ML Potentials: A Practical Guide for Molecular Dynamics in Biomedical Research
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on deploying Machine Learning Potentials (MLPs) within the LAMMPS molecular dynamics framework.
Beyond Schrödinger: Understanding the Kohn-Sham Equations for Modern Drug Discovery
This article provides a comprehensive guide to the Kohn-Sham (KS) equations, the cornerstone of modern Density Functional Theory (DFT), tailored for computational researchers in biomedical science and drug development.
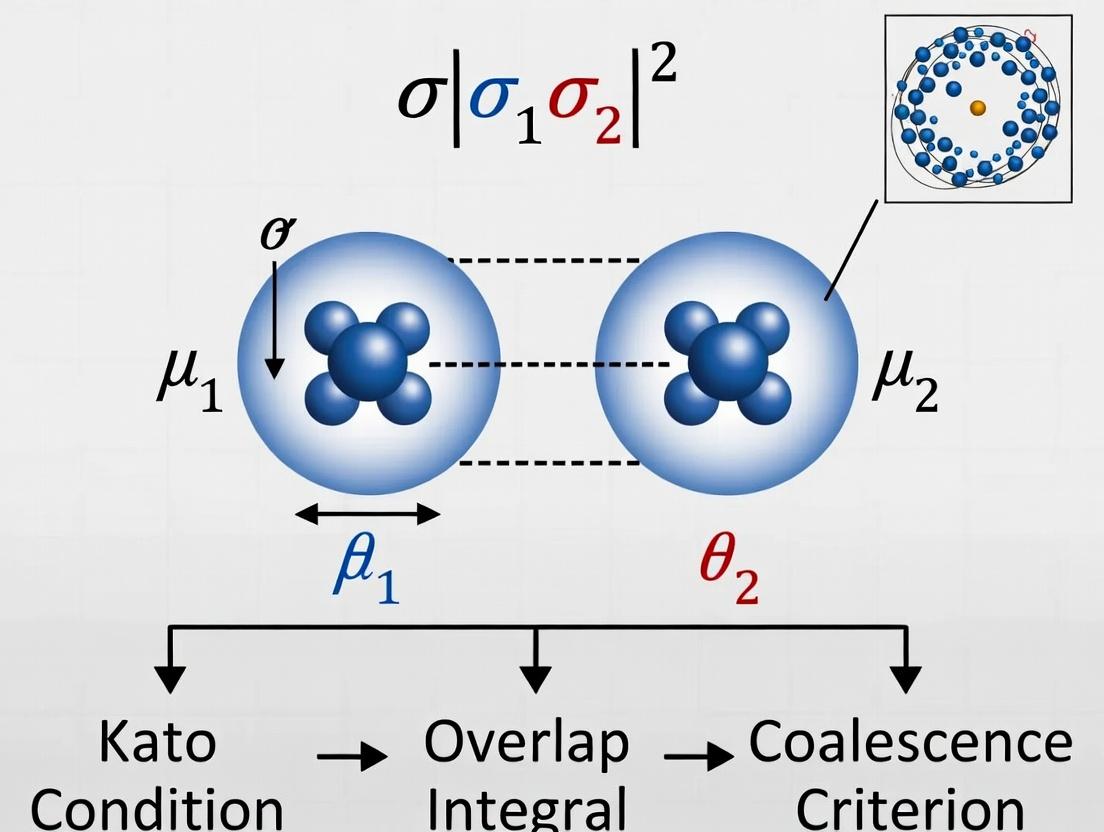
The Kato Cusp Condition Explained: A Quantum Mechanical Bridge from Electron Coalescence to Biomolecular Accuracy
This article provides a comprehensive exploration of the Kato cusp condition, a fundamental constraint in quantum mechanics governing the behavior of wavefunctions at points of electron-electron coalescence.