AI and Quantum Chemistry: Computational Approaches for Mapping Chemical Reaction Mechanisms in Drug Discovery
This article explores the transformative role of computational chemistry in elucidating chemical reaction mechanisms, a cornerstone of modern drug discovery and development.
AI and Quantum Chemistry: Computational Approaches for Mapping Chemical Reaction Mechanisms in Drug Discovery
Abstract
This article explores the transformative role of computational chemistry in elucidating chemical reaction mechanisms, a cornerstone of modern drug discovery and development. We examine foundational principles, from quantum mechanics to generative AI, that enable the prediction of reaction pathways and transition states. The scope encompasses a detailed review of cutting-edge methodologies—including machine learning potentials, large language model-guided exploration, and ultra-large virtual screening—and their practical applications in pharmaceutical research. The content further addresses troubleshooting computational limitations and provides a comparative analysis of tool validation, offering researchers and drug development professionals a comprehensive guide to leveraging these technologies for accelerating the design of novel therapeutics.
From Electrons to Outcomes: The Physical Principles of Reaction Prediction
The discovery and optimisation of novel small-molecule drug candidates critically hinges on the efficiency of the iterative Design-Make-Test-Analyse (DMTA) cycle [1]. Within this framework, the synthesis ("Make") phase consistently represents the most costly and time-consuming element, often creating a significant bottleneck that slows drug development pipelines [1]. This challenge intensifies when targeting complex biological systems, which frequently demand intricate chemical structures that require multi-step synthetic routes. These routes are inherently labour-intensive, involving numerous variables that must be scouted and optimised before a successful pathway is identified [1]. The core of this "Make" bottleneck lies in the fundamental difficulty of accurately predicting reaction outcomes—including yield, regioselectivity, and stereochemistry—before compounds are ever synthesised in the laboratory. Overcoming this predictive challenge is not merely a technical improvement but a crucial requirement for accelerating the delivery of new therapeutics to patients.
The High Stakes of Reaction Prediction
Impact on the Drug Discovery Workflow
Inaccurate reaction prediction has direct and severe consequences on drug discovery efficiency. When synthesis fails or yields an unexpected product, the result is wasted resources, extended timelines, and ultimately, a limitation on the chemical space that can be feasibly explored for potential drug candidates [1]. The DMTA cycle relies on the rapid and reliable synthesis of compound series for biological evaluation. Any failure to obtain the desired chemical matter for testing invalidates the entire iterative process, stalling projects and consuming substantial financial and human resources that could be allocated elsewhere [1]. Furthermore, the explorable chemical space is directly dictated by the available building blocks and the confidence with which they can be combined into novel molecular architectures [1]. Without reliable prediction, chemists must resort to conservative, well-established reactions, potentially missing superior drug candidates that reside in less-charted chemical territory.
Quantitative Evidence of the Prediction Challenge
Experimental data and model performance metrics underscore the magnitude of the prediction challenge. The following table summarises key quantitative evidence from recent studies:
Table 1: Quantitative Evidence of Reaction Prediction Challenges
| Evidence Type | Description | Impact/Performance | Source |
|---|---|---|---|
| Model Accuracy | Molecular Transformer Top-1 accuracy on standard USPTO dataset | 90% (biased split) | [2] |
| Model Accuracy | Molecular Transformer Top-1 accuracy on debiased dataset | Significant decrease (exact % not stated) | [2] |
| Reaction Class Failure | Diels–Alder reaction prediction | Inability to predict regioselectivity; wrong product predicted | [2] |
| Data Scarcity | Diels–Alder reactions in USPTO training data | Very few instances, explaining poor performance | [2] |
| Condition Prediction | GraphRXN model on in-house HTE Buchwald-Hartwig data | R² = 0.712 for yield prediction | [3] |
The performance drop observed when moving from a standard to a debiased dataset is particularly revealing. It indicates that reported high accuracies can be inflated by "Clever Hans" predictions, where models make correct predictions for the wrong reasons due to hidden biases in the data, such as spurious correlations between specific substituents and outcomes [2]. This phenomenon masks fundamental shortcomings in model generalisability.
Technical Hurdles in Reaction Outcome Prediction
Data Limitations and Biases
The development of robust predictive models is severely constrained by data availability and quality. Most reaction data available in public databases and literature suffer from a pronounced positive results bias, where failed reactions are systematically underreported [3]. This creates an incomplete picture of chemical reactivity, as models never learn what does not work. Furthermore, dataset scaffold bias—where certain molecular frameworks are overrepresented—leads to models that perform well on familiar scaffolds but fail on novel ones [2]. Finally, there is a pervasive issue of incomplete annotation, where crucial contextual metadata like reaction temperature, scale, or the scientific focus of the project (e.g., medicinal chemistry vs. total synthesis) is omitted [2]. Without this context, which a skilled chemist intuitively uses to interpret reactions, models struggle to make reliable predictions.
Mechanistic Complexity and Computational Cost
The fundamental challenge in reaction prediction lies in accurately modelling the Potential Energy Surface (PES), which depicts the energy states associated with atomic positions during a chemical transformation [4]. Understanding reaction kinetics and feasibility hinges on identifying key points on this surface: reactants and intermediates (energy minima) and transition states (first-order saddle points connecting them) [4]. Transition states are particularly elusive, as they are transient and typically must be revealed through theoretical simulations. Exploring the PES for complex, multi-step reactions presents a combinatorial explosion of possible pathways, making exhaustive searches computationally prohibitive [4]. While high-level quantum mechanical (QM) methods like Density Functional Theory (DFT) offer accuracy, they are notoriously time-intensive and resource-heavy, limiting their application for high-throughput screening in large reaction spaces [4] [3].
The Interpretability Gap in Machine Learning Models
While machine learning models, particularly deep learning architectures, show promise in reaction prediction, their "black-box" nature presents a significant hurdle to adoption by chemists. For model users, the inability to understand why a model predicts a particular outcome is problematic because chemical reactions are highly contextual [2]. A prediction lacking a chemically rational explanation is of limited utility for making strategic decisions in a synthesis campaign. For model developers, this opaqueness makes it difficult to diagnose failure modes and improve model design. It remains unclear whether state-of-the-art models like the Molecular Transformer are learning true physicochemical principles or merely exploiting superficial statistical patterns in the training data [2]. This interpretability gap hinders trust and the effective integration of AI tools into the medicinal chemist's workflow.
Methodologies and Experimental Protocols
AI and Machine Learning Approaches
A. Transformer-Based Architectures: The Molecular Transformer adapts the neural machine translation architecture to chemistry, treating reaction prediction as a translation task where reactant and reagent SMILES strings are "translated" into product SMILES strings [2]. The model relies on a self-attention mechanism to weigh the importance of different parts of the input molecules when generating the output.
Protocol for Training a Molecular Transformer Model
- Data Preprocessing: A large dataset of reactions (e.g., the USPTO dataset text-mined from patents) is collected. SMILES strings of reactants, reagents, and products are canonicalised and combined into a single sequence using a special token (e.g., ">") to separate reactants from reagents.
- Data Augmentation: The model's robustness is improved by employing SMILES augmentation, where each reaction is represented multiple times using different, equivalent SMILES string permutations [2].
- Model Training: The transformer model, comprising an encoder and decoder stack, is trained to learn the mapping from input sequences (reactants + reagents) to output sequences (products). Training involves minimising the cross-entropy loss between the predicted and actual product sequences.
- Interpretation: Post-training, interpretation techniques like Integrated Gradients (IG) are used to attribute the model's predictions to specific substructures in the input. This helps validate whether the model is learning chemically sensible features [2].
B. Graph Neural Networks (GNNs): Frameworks like GraphRXN represent molecules as graphs, where atoms are nodes and bonds are edges [3]. These models directly learn from 2D molecular structures.
Protocol for the GraphRXN Framework
- Graph Representation: Each reaction component (reactant, reagent, solvent) is converted into a directed molecular graph G(V, E), where V is the set of nodes (atoms) and E is the set of edges (bonds). Node and edge features are initialised based on atom and bond types.
- Message Passing: A modified communicative message passing neural network is used. Over K steps, nodes and edges iteratively update their hidden states by aggregating information from their local neighbourhoods.
- For a node v, its message vector is aggregated from its connected edges.
- For an edge e_{v,w}, its message is derived from the hidden states of its source node and the edge itself.
- Readout: After K iterations, a Gated Recurrent Unit (GRU) aggregates the final node embeddings into a single, fixed-length molecular feature vector for each reaction component.
- Reaction-Level Prediction: The molecular vectors of all components are aggregated (e.g., via summation or concatenation) into a single reaction vector. This vector is fed into a dense neural network to predict the reaction output, such as reaction success or yield [3].
C. Hybrid and Rule-Guided Approaches: Tools like ARplorer and RxnNet integrate quantum mechanics with rule-based methodologies or heuristic chemical knowledge to explore reaction pathways more efficiently [4] [5].
Protocol for ARplorer's Reaction Pathway Exploration
- Active Site Identification: The program identifies potential reactive sites and bond-breaking locations in the input molecular structures.
- LLM-Guided Chemical Logic: A Large Language Model (LLM), primed with general chemical knowledge from literature and system-specific rules based on functional groups, generates chemical logic and SMARTS patterns to guide the search and filter unlikely pathways [4].
- Structure Optimisation & TS Search: An iterative process optimises molecular structures and searches for transition states. This employs an active-learning sampling method to enhance efficiency.
- IRC Analysis & Pathway Finalisation: Intrinsic Reaction Coordinate (IRC) calculations are performed from located transition states to connect reactants, intermediates, and products. Duplicate pathways are removed, and the final reaction network is constructed [4].
The following diagram illustrates the hybrid workflow of the ARplorer program, which combines quantum mechanics with LLM-guided chemical logic for efficient reaction pathway exploration.
High-Throughput Experimentation (HTE) for Data Generation
To address the critical lack of high-quality, unbiased reaction data, High-Throughput Experimentation (HTE) has emerged as a powerful experimental protocol [3]. HTE enables the systematic and parallel execution of hundreds or thousands of reactions under varying conditions.
Protocol for Generating HTE Datasets
- Reaction Selection: A reaction of interest (e.g., Buchwald-Hartwig amination, Suzuki coupling) is selected.
- Experimental Design: A diverse set of substrates, catalysts, ligands, solvents, and bases is chosen to create a broad matrix of reaction conditions.
- Automated Execution: Reactions are set up robotically in miniaturised format (e.g., in well-plates) to ensure consistency and reproducibility.
- Analysis & Data Curation: Reaction outcomes (e.g., yield, conversion) are analysed using high-throughput analytics (e.g., UPLC-MS). All results—both successful and failed—are recorded in a standardised format with complete metadata. This creates a balanced dataset ideal for training machine learning models [3].
The following table details key computational and experimental reagents essential for research in reaction outcome prediction.
Table 2: Research Reagent Solutions for Predictive Synthesis
| Tool/Resource | Type | Primary Function | Example/Standard |
|---|---|---|---|
| CASP Tools | Software | Computer-Assisted Synthesis Planning for retrosynthetic analysis and route design | AI-powered platforms (e.g., from Roche, Molecular Transformer) [1] |
| HTE Platforms | Experimental | Robotic systems for high-throughput, parallelised reaction execution and data generation | Custom/in-house platforms; commercial systems [3] |
| FAIR Data Repositories | Data | Stores for Findable, Accessible, Interoperable, and Reusable reaction data | Internal corporate databases; public databases (e.g., USPTO) [1] |
| Building Block Catalogues | Chemical | Sources of diverse starting materials (BBs) for synthesis | Enamine, eMolecules, Sigma-Aldrich; virtual MADE catalogue [1] |
| QM Software | Software | Performs quantum chemical calculations to explore Potential Energy Surfaces | Gaussian, GFN2-xTB [4] |
| Reaction Fingerprints | Computational | Numerical representation of reactions for machine learning modelling | DRFP, MFFs, Graph-based learned representations (GraphRXN) [3] |
Integrated Workflows and Future Directions
The true power of predictive synthesis is realised when these methodologies are combined into integrated, data-driven workflows. The future lies in closing the loop between in-silico prediction and automated experimental validation. A promising workflow begins with AI-powered synthesis planning, which generates proposed routes for a target molecule [1]. These proposals are then vetted by a medicinal chemist, potentially interacting with a "Chemical ChatBot" in an iterative dialogue to refine the plan [1]. The most promising routes are executed on automated synthesis platforms, which generate high-quality FAIR (Findable, Accessible, Interoperable, and Reusable) data [1]. This data is fed back into the AI models, continuously refining their predictive capabilities and creating a self-improving cycle.
The diagram below illustrates this envisioned, fully integrated workflow for data-driven synthesis, highlighting the seamless connection between design, AI planning, automated execution, and data analysis.
Key future developments include the deeper integration of LLMs as interfaces to complex models and data, the move towards unified models that simultaneously predict retrosynthetic pathways and reaction conditions, and a cultural shift towards treating high-quality data stewardship as a central pillar of chemical research [1] [4]. As these trends converge, the ability to predict reaction outcomes with high fidelity will cease to be a central challenge and instead become a cornerstone of a accelerated, more efficient drug discovery process.
The computational exploration of chemical reaction mechanisms represents a cornerstone of modern research, driving advances in fields from drug development to materials science. As artificial intelligence (AI) and machine learning (ML) models assume increasingly prominent roles in these explorations, their predictions must adhere to the fundamental laws of physics to be scientifically trustworthy. Among these laws, the principles of mass conservation and electron conservation are non-negotiable; they form the foundational reality upon which all chemical processes occur. Mass conservation states that for any system closed to matter transfer, the mass must remain constant over time, meaning atoms can be rearranged but neither created nor destroyed [6]. Similarly, electron conservation is critical for modeling redox processes and electronic interactions accurately. Unfortunately, many data-driven models, including sophisticated large language models, often violate these core tenets, producing predictions that are physically impossible and thus of limited utility for rigorous scientific inquiry [7] [8]. This whitepaper details the critical importance of embedding these conservation laws as hard constraints within AI frameworks, surveys current methodological approaches, and provides practical protocols for researchers seeking to develop physically-grounded models for computational chemistry.
Mathematical and Physical Foundations of Conservation Laws
The Principle of Mass Conservation
The law of conservation of mass is a bedrock principle in chemistry and physics. Formally, for a closed system, the total mass of reactants must equal the total mass of products in any chemical reaction [9] [6]. This principle emerged from centuries of scientific inquiry, with Antoine Lavoisier's meticulous experiments in the late 18th century definitively demonstrating that although substances may change form during reactions, their total mass remains invariant [6]. Mathematically, for a chemical system with m compounds formed from p elements, this conservation can be expressed as:
M^T ΔC = 0_p
where M is the m × p composition matrix (containing the atomic composition of each species), ΔC is the vector of concentration changes for each species, and 0_p is a zero vector of length p [7]. This equation ensures that the total number of atoms of each element remains constant throughout any transformation.
The Critical Need for Electron Conservation
While mass conservation provides a macroscopic constraint, electron conservation operates at the quantum mechanical level and is equally vital for modeling chemical reactivity accurately. Electrons are the currency of chemical bonds, and their redistribution dictates reaction pathways. The challenge of electron conservation is particularly acute in AI models that attempt to predict reaction outcomes, as standard models may artificially create or annihilate electrons, leading to unrealistic predictions [8]. One promising approach to this challenge utilizes a bond-electron matrix, a concept dating back to Ivar Ugi's work in the 1970s, which represents the electrons involved in a reaction explicitly. This matrix uses nonzero values to represent bonds or lone electron pairs and zeros elsewhere, providing a framework that simultaneously conserves both atoms and electrons [8].
Current AI Approaches and Their Limitations
The Conservation Challenge in Machine Learning
Many machine learning applications in chemistry operate as "black boxes" that learn patterns from data but lack built-in mechanisms to enforce physical laws. This is particularly true for large language models (LLMs) adapted for chemical prediction tasks. As noted by MIT researchers, when these models use computational "tokens" representing individual atoms without conservation constraints, "the LLM model starts to make new atoms, or deletes atoms in the reaction," resulting in predictions that resemble "a kind of alchemy" rather than scientifically grounded chemistry [8]. Similar issues plague models predicting atmospheric composition, where unphysical deviations from mass conservation, though sometimes minor, undermine the models' scientific credibility [7].
Promising Approaches for Physically-Grounded AI
Several innovative approaches are emerging to address these limitations by embedding physical constraints directly into AI frameworks:
Projection-Based Nudging: This method takes the output of any numerical model and minimally adjusts the predicted concentrations to the nearest physically consistent solution that respects atomic conservation laws. The correction uses a single matrix operation derived from constrained optimization theory, projecting predictions onto the null space of the composition matrix
M^Tto ensure mass conservation to machine precision [7].Flow Matching for Electron Redistribution (FlowER): Developed at MIT, this generative AI approach uses a bond-electron matrix to explicitly track all electrons in a reaction, ensuring none are spuriously added or deleted. The system shows promising results for predicting realistic mechanistic pathways while maintaining real-world physical constraints [8].
Heuristics-Guided Exploration: This computational protocol constructs reaction networks using heuristic rules derived from conceptual electronic-structure theory while ensuring conservation through quantum chemical optimization of generated structures [10].
Table 1: Comparison of AI Approaches with Physical Constraints
| Method | Conservation Principle | Key Mechanism | Reported Advantages |
|---|---|---|---|
| Projection-Based Nudging [7] | Mass/Atom Conservation | Matrix-based projection to nearest physical solution | Model-agnostic, minimal perturbation, closed-form solution |
| FlowER [8] | Mass & Electron Conservation | Bond-electron matrix representation | Realistic reaction predictions, maintains electronic constraints |
| Heuristics-Guided Exploration [10] | Implicit via QM optimization | Structure generation based on chemical rules | Automated discovery of reaction pathways |
| Trajectory-Based Methods (tsscds) [11] | Implicit via QM methods | Accelerated molecular dynamics and graph theory | Discovers mechanisms with minimal human intervention |
Practical Implementation and Experimental Protocols
Implementing Mass Conservation as a Hard Constraint
For researchers implementing mass conservation in existing models, the projection-based nudging method provides a practical, post-hoc correction. The protocol involves:
Define the Composition Matrix: Construct the
m × pcomposition matrixMwhere entries represent the number of atoms of each elementpin each chemical speciesm.Compute the Correction Matrix: Calculate the projection matrix
M_corrusing the formula:M_corr = I - M(M^T M)^-1 M^TwhereIis the identity matrix [7].Apply the Correction: For any model prediction
ΔC'(representing concentration changes or tendencies), the mass-conserving solution is obtained as:ΔC = M_corr × ΔC'Species-Weighted Extension: For systems with varying uncertainty across species, implement a weighted version that considers the uncertainty and magnitude of each species, preferentially adjusting species with lower predicted accuracy [7].
Workflow for Electron-Conserving Reaction Prediction
The FlowER protocol for predicting chemical reactions while conserving electrons involves:
Representation: Convert molecular structures into a bond-electron matrix that explicitly represents bonds and lone electron pairs.
Flow Matching: Employ flow matching techniques to model the redistribution of electrons throughout the reaction process.
Constraint Enforcement: Maintain nonzero values in the matrix to represent bonds or lone electron pairs and zeros to represent their absence, ensuring conservation of both atoms and electrons throughout the transformation [8].
The following diagram illustrates a comprehensive workflow for implementing these conservation principles in AI-driven reaction exploration:
Research Reagents and Computational Tools
Table 2: Essential Research Reagents and Computational Tools for Conservation-Grounded AI
| Tool/Reagent | Type | Function/Purpose | Implementation Notes |
|---|---|---|---|
| Composition Matrix (M) [7] | Mathematical Framework | Encodes elemental composition of all species | Foundation for mass conservation constraints |
| Bond-Electron Matrix [8] | Representation Scheme | Tracks electrons and bonds explicitly | Ensures electron conservation in reactions |
| Projection Matrix (M_corr) [7] | Computational Operator | Nudges predictions to mass-conserving solutions | Can be weighted by species uncertainty |
| Graph Theory Algorithms [11] | Analysis Tool | Identifies reaction pathways and connectivity | Uses adjacency matrices to track bond changes |
| Quantum Chemistry Codes [12] | Validation Tool | Provides benchmark energies and structures | DFT, coupled-cluster, or semiempirical methods |
| Automated Exploration Software [13] | Discovery Platform | Systematically explores reaction mechanisms | Tools like CHEMOTON, SCINE, tsscds |
Advanced Exploration and Steering of Chemical Reaction Networks
For complex chemical systems, particularly in catalysis and drug discovery, merely predicting single reactions is insufficient. Researchers need tools to explore entire reaction networks while maintaining physical constraints. The STEERING WHEEL algorithm addresses this challenge by providing human-machine collaboration for exploring chemical reaction networks [13]. This approach alternates between network expansion steps (which add new calculations and results to a growing reaction network) and selection steps (which choose subsets of structures to limit combinatorial explosion) [13]. The following diagram illustrates this interactive exploration process:
This guided approach is particularly valuable for transition metal catalysis and complex organic transformations, where the reaction space is vast and a brute-force exploration is computationally unfeasible [13]. By combining human chemical intuition with automated exploration, researchers can efficiently map out relevant regions of chemical space while maintaining physical constraints throughout the process.
The integration of mass and electron conservation principles into AI frameworks for chemical prediction is not merely a theoretical enhancement but a practical necessity for producing scientifically valid results. As computational chemistry continues to embrace data-driven methods, the fundamental laws of physics must serve as the immutable foundation upon which these models are built. The methodologies surveyed here—from projection-based nudging to electron-conserving generative models and guided network exploration—provide researchers with powerful tools to ensure their AI systems remain grounded in physical reality. For drug development professionals, materials scientists, and chemical researchers, adopting these constraint-based approaches is critical for accelerating discovery while maintaining scientific rigor in the computational exploration of chemical reaction mechanisms.
The accurate prediction of chemical reaction outcomes represents a cornerstone of modern chemical research, with profound implications for drug discovery, materials science, and sustainable chemical synthesis. For decades, computational chemists have sought to develop models that can reliably forecast the products and pathways of chemical transformations. However, many data-driven approaches have struggled with a fundamental limitation: their inability to consistently obey the laws of physics, particularly the conservation of mass and electrons. This violation of physical constraints has resulted in what researchers term "hallucinatory failure modes," where models predict chemically impossible structures with atoms appearing or disappearing spontaneously [14] [8]. Such limitations have restricted the practical utility of computational tools in real-world discovery pipelines.
The recent introduction of FlowER (Flow matching for Electron Redistribution) by MIT researchers represents a paradigm shift in this landscape. By grounding predictions in the physical reality of electron movement through bond-electron matrices, this approach enforces strict conservation laws while maintaining predictive accuracy [14] [8] [15]. This technical guide examines the core innovations of this methodology, its experimental validation, and its implications for computational exploration of chemical reaction mechanisms.
Core Innovation: Bond-Electron Matrix Representation
Theoretical Foundation and Historical Context
The bond-electron (BE) matrix framework employed in FlowER has its roots in work from the 1970s by chemist Ivar Ugi, who developed this representation to systematically track electrons in chemical systems [8] [16]. This approach encodes atomic identities and their electron configurations in a compact matrix format where:
- Nonzero values represent bonds or lone electron pairs
- Zeros indicate the absence of bonding or lone pair electrons [8] [17]
This mathematical representation naturally embeds two critical conservation principles directly into the model's architecture: (1) conservation of all atoms and (2) conservation of all electrons [14]. The BE matrix directly reflects the conventions of arrow-pushing diagrams that chemists have used for generations to visualize reaction mechanisms, creating a bridge between traditional chemical intuition and modern machine learning approaches [14].
From String Transformation to Electron Redistribution
Traditional sequence-based models treat chemical reactions as string transformations—converting reactant SMILES strings to product SMILES strings through pattern recognition [16] [15]. This approach fundamentally disregards the physical entities underlying the transformations. As researchers noted, "if you don't conserve the tokens, the LLM model starts to make new atoms, or deletes atoms in the reaction," resulting in predictions that resemble "alchemy" rather than scientifically grounded chemistry [8].
FlowER recasts this problem entirely by modeling chemical reactivity as "a generative process of electron redistribution" [14] [18]. Instead of treating atoms as tokens in a string, the system explicitly tracks electron movement throughout the reaction process, ensuring that predictions align with physical reality [14]. The matrix representation enables this by providing a complete description of covalent bonding and lone pairs at any pseudo-timepoint between reactants (t=0) and products (t=1) [14].
Table 1: Comparison of Chemical Reaction Representation Approaches
| Representation | Fundamental Unit | Conservation Enforcement | Interpretability |
|---|---|---|---|
| SMILES Strings | Character tokens | None inherent; frequently violated | Low; black-box transformation |
| Graph Edits | Bond changes | Partial; often atom-only | Medium; shows bond changes but not electron movement |
| Bond-Electron Matrix | Electrons and atoms | Built-in to architecture; exact conservation | High; aligns with arrow-pushing mechanisms |
FlowER Architecture: Implementing Physical Constraints
Flow Matching Framework
FlowER employs a modern deep generative framework called flow matching, which generalizes diffusion-based approaches while offering faster inference [14]. This framework formalizes electron movement as the transformation of a probability distribution of electron localization from the reactants' state to the products' state [14]. The model learns to analyze any intermediate state between reactants and products by featurizing the BE matrix and atom identities at pseudo-timepoints between t=0 (reactants) and t=1 (products) [14].
At its core, FlowER utilizes a graph transformer architecture with a multi-headed attention mechanism that operates on the BE matrix representation [14]. The model predicts electron movements analogous to partial arrow-pushing diagrams, which are then applied to update the BE matrix for subsequent timepoints. This recursive prediction yields a complete reaction mechanism step-by-step while ensuring each intermediate state adheres to strict conservation principles [14].
The ΔBE Matrix and Electron Conservation
The central prediction of FlowER is the ΔBE matrix, which captures changes in electron configurations with a net sum of zero, thereby enforcing exact electron conservation [14]. This approach directly reflects the conventions of arrow-pushing diagrams, providing predictions that align with how chemists visualize and rationalize reaction mechanisms [14]. The model further distinguishes between lone pair and bond electron distributions, capturing the nuanced roles of electrons in chemical bonding and reactivity [14].
The following diagram illustrates the core workflow of the FlowER model for predicting reaction mechanisms through electron redistribution:
Diagram 1: FlowER model workflow for electron-conserving reaction prediction
Experimental Protocol and Validation
Training Methodology and Dataset Curation
To train FlowER, the research team imputed mechanistic pathways for a subset of the USPTO-Full dataset containing approximately 1.1 million experimentally-demonstrated reactions from United States Patent Office patents [14]. This comprehensive dataset was processed using 1,220 expert-curated reaction templates constructed for 252 well-described reaction classes, resulting in a total of 1.4 million elementary reaction steps [14].
Following the standard training procedure for conditional flow matching, the team used interpolative trajectories sampled between reactant and product BE matrices as input, with the difference in reactant-product BE matrices serving as ground truth during model training [14]. This approach allowed the model to learn the continuous process of electron redistribution while maintaining physical constraints throughout the transformation.
Research Reagent Solutions
Table 2: Essential Research Components for FlowER Implementation
| Component | Function | Implementation Details |
|---|---|---|
| USPTO-Full Dataset | Training data source | ~1.1 million patented reactions providing experimental validation [14] |
| Bond-Electron Matrix | Physical representation | Encodes atoms, bonds, and lone pairs while enforcing conservation [14] [8] |
| Graph Transformer | Neural architecture | Processes BE matrix with multi-headed attention [14] |
| Flow Matching | Generative framework | Models probability path from reactants to products [14] |
| Mechanistic Templates | Reaction classification | 1,220 expert-curated templates across 252 reaction classes [14] |
Quantitative Performance Analysis
Conservation Law Adherence
The most significant advantage of FlowER's bond-electron matrix approach appears in its strict adherence to conservation laws. When evaluated at the single elementary step level, FlowER demonstrated remarkable performance compared to sequence-based models:
Table 3: Conservation Law Adherence in Reaction Prediction Models
| Model | Valid SMILES | Heavy Atom Conservation | Full Mass & Electron Conservation |
|---|---|---|---|
| FlowER | ~95% | Enforced by architecture | Enforced by architecture |
| Graph2SMILES (G2S) | 68.9% | 31.4% | 14.3% |
| Graph2SMILES+H | 77.28% | 30.1% | 17.3% |
The data reveal that despite being trained on balanced mechanistic datasets, sequence generative models violate fundamental conservation laws for the majority of predictions [14]. Only 14.3-17.3% of sequence-based predictions maintained complete conservation of heavy atoms, protons, and electrons, compared to FlowER's architectural enforcement of these fundamental constraints [14].
Generalization and Data Efficiency
Beyond conservation, FlowER demonstrates impressive generalization capabilities, recovering complete mechanistic sequences with strict mass conservation and learning fundamental chemical principles that connect to expert intuition [14]. The model's physical grounding enables downstream thermodynamic evaluations of reaction feasibility, providing insights beyond mere structural prediction [14].
Perhaps most notably, FlowER achieves remarkable fine-tuning performance on unseen reaction classes with only 32 reaction examples, demonstrating unprecedented sample efficiency for a chemical prediction model [14]. This data-efficient generalization suggests that the model internalizes chemical principles rather than merely memorizing reaction patterns.
Implications for Computational Reaction Mechanism Research
Bridging Mechanistic Understanding and Prediction
FlowER represents a significant advancement in bridging the gap between predictive accuracy and mechanistic understanding in data-driven reaction outcome prediction [14]. By providing explicit electron redistribution pathways alongside product predictions, the model offers interpretable insights that align with chemical intuition [15]. This dual capability addresses a longstanding criticism of "black box" AI models in chemistry, which often fail to explain why a particular product is predicted [14].
The model's probabilistic nature also enables exploration of branching mechanistic pathways, side products, and potential impurities through repeated sampling—a capability that mirrors the reality of chemical systems where multiple pathways often compete [14]. This represents a departure from deterministic prediction models that typically identify only the single most likely outcome.
Limitations and Future Directions
The MIT team has been transparent about FlowER's current limitations, particularly its restricted coverage of reactions involving metals and catalytic cycles [8] [17]. These gaps stem from the training data sourced from patent literature, which contains limited examples of these chemistries [8]. Expansion to encompass organometallic chemistry, catalysis, and electrochemical systems represents an important direction for future development.
Additionally, while the physics-grounded approach is elegant, it also increases model complexity compared to simpler pattern-matching approaches [16]. The scalability of this approach to the vastness of chemical space remains an open question, though the demonstrated sample efficiency in fine-tuning suggests promising extensibility [14] [16].
The bond-electron matrix approach implemented in FlowER represents a fundamental shift in how computational models conceptualize and predict chemical reactivity. By embedding physical constraints directly into the model architecture rather than treating them as optional guidelines, this methodology addresses core limitations that have plagued data-driven chemistry models for decades. The result is a system that not only predicts reaction outcomes but does so through mechanistically interpretable pathways that obey the fundamental laws of chemistry and physics.
As the field progresses toward more sophisticated AI-assisted chemical discovery, approaches like FlowER that prioritize physical realism alongside predictive accuracy will be essential for building trust and utility in real-world applications. The integration of physical principles with data-driven learning represents a promising path toward computational tools that truly understand chemistry rather than merely mimicking its patterns.
The Schrödinger equation serves as the fundamental cornerstone of quantum mechanics, providing the mathematical framework necessary for describing and predicting the behavior of particles at the atomic and subatomic levels. In computational chemistry, this equation enables researchers to move beyond observational chemistry to predictive, first-principles calculations of molecular structure, properties, and reactivity. This technical guide explores the essential role of the Schrödinger equation in the computational exploration of chemical reaction mechanisms, with particular relevance to pharmaceutical research and drug development. By establishing the theoretical foundation and presenting practical methodologies, this work aims to equip researchers with the knowledge to leverage quantum chemical computations in mechanistic studies.
The Schrödinger equation is a partial differential equation that forms the quantum counterpart to Newton's second law in classical mechanics [19]. Named after Erwin Schrödinger who postulated it in 1926, this equation describes how the quantum state of a physical system changes over time [20] [19]. Unlike Newtonian mechanics which predicts definite paths for particles, the Schrödinger equation operates on the wave function, denoted as |Ψ⟩, which contains all the information about a quantum system [19].
In the context of computational chemistry, the time-independent Schrödinger equation is of particular importance for determining the stable states of molecular systems [20]. This formulation appears as an eigenvalue equation:
Ĥ|ψ⟩ = E|ψ⟩
Where Ĥ is the Hamiltonian operator representing the total energy of the system, |ψ⟩ is the wave function of the system, and E is the energy eigenvalue corresponding to that particular state [20]. Solving this equation for a chemical system provides the allowable energy states and electron distributions, which directly determine molecular properties and reactivity [20].
The linearity of the Schrödinger equation is a crucial mathematical property with profound physical implications [19]. If |ψ₁⟩ and |ψ₂⟩ are both possible states of a system, then any linear combination |ψ⟩ = a|ψ₁⟩ + b|ψ₂⟩ is also a valid state [20] [19]. This principle of superposition enables quantum systems to exist in multiple states simultaneously, a phenomenon with significant consequences for molecular behavior and quantum computing applications in chemistry [20].
Mathematical Foundation
Fundamental Forms and Operators
The Schrödinger equation exists in two primary forms: time-dependent and time-independent. The time-dependent Schrödinger equation governs the evolution of quantum systems:
iℏ(∂/∂t)|Ψ(t)⟩ = Ĥ|Ψ(t)⟩
Here, i is the imaginary unit (√-1), ℏ is the reduced Planck constant, and Ĥ is the Hamiltonian operator [19]. For many practical applications in computational chemistry, the time-independent form is more directly useful:
Ĥ|ψₙ⟩ = Eₙ|ψₙ⟩
This eigenvalue equation provides stationary states of the system, where |ψₙ⟩ represents the wave function of the nth stationary state and Eₙ is its corresponding energy [19].
The Hamiltonian operator Ĥ encapsulates the total energy of the system and consists of two fundamental components [20]:
Ĥ = T̂ + V̂
Where T̂ represents the kinetic energy operator and V̂ represents the potential energy operator. For a single particle in one dimension, the kinetic energy operator takes the form -ℏ²/2m(∂²/∂x²), while the potential energy operator V(x) depends on the specific system being studied [19].
Table 1: Key Components of the Schrödinger Equation
| Component | Mathematical Representation | Physical Significance | ||
|---|---|---|---|---|
| Wave Function | Ψ⟩ or Ψ(x,t) | Complete description of quantum state; contains all measurable information | ||
| Hamiltonian Operator | Ĥ = T̂ + V̂ | Total energy of the system | ||
| Kinetic Energy Operator | -ℏ²/2m(∂²/∂x²) | Energy due to particle motion | ||
| Potential Energy Operator | V(x,t) | Energy from interactions and external fields | ||
| Probability Density | Ψ(x,t) | ² | Probability of finding particle at position x at time t |
Physical Interpretation and Observables
The wave function solution to the Schrödinger equation, Ψ(x,t), has a probabilistic interpretation first proposed by Max Born [19]. Specifically, the square of the absolute value of the wave function, |Ψ(x,t)|², defines a probability density function. For a wave function in position space, this means:
Pr(x,t) = |Ψ(x,t)|²
This equation indicates the probability of finding the particle at position x at time t [19]. This probabilistic nature fundamentally distinguishes quantum mechanics from classical physics.
When the Schrödinger equation is solved for a system, the resulting wave functions represent stationary states with precisely defined energies [20]. These solutions represent the only allowed energy states for the system, which has profound implications for molecular structure and spectroscopy. The quantum superposition principle allows general states to be constructed as linear combinations of these energy eigenstates [20] [19].
The Schrödinger Equation in Computational Chemistry
From Mathematical Formalism to Chemical Insight
In computational chemistry, the Schrödinger equation provides the theoretical foundation for understanding and predicting chemical phenomena at the most fundamental level. The process begins with constructing the molecular Hamiltonian, which incorporates the kinetic energies of all electrons and nuclei, as well as the potential energy terms describing all Coulombic interactions between these charged particles [20].
The complexity of solving the Schrödinger equation increases dramatically with system size [20]. For a hydrogen atom (one electron), an exact solution is possible, but for larger atoms and molecules, the electron-electron repulsion terms make analytical solutions intractable [20]. This challenge has driven the development of sophisticated computational methods, including the Hartree-Fock approach, density functional theory, and quantum Monte Carlo techniques, all of which represent different strategies for approximating solutions to the Schrödinger equation for many-electron systems.
The power of these computational approaches lies in their ability to extract chemically meaningful information from the wave function. For example, the electron density derived from the wave function can be visualized to reveal molecular orbitals, bond critical points, and reaction pathways. Additionally, the energy eigenvalues provide access to thermodynamic properties, while the response of the wave function to external perturbations enables prediction of spectroscopic parameters.
Computational Workflow for Reaction Mechanism Exploration
The application of quantum chemistry to reaction mechanism exploration follows a systematic workflow that transforms molecular structures into mechanistic insights. The diagram below illustrates this process:
Diagram 1: Quantum Chemistry Workflow
Key Methodologies and Protocols
Computational exploration of reaction mechanisms relies on several well-established protocols built upon the Schrödinger equation foundation. The table below summarizes the primary computational methodologies:
Table 2: Computational Methodologies for Reaction Mechanism Studies
| Methodology | Theoretical Basis | Key Applications in Mechanism Research | Computational Cost |
|---|---|---|---|
| Ab Initio Methods | Direct solution of electronic Schrödinger equation with approximate wave function | High-accuracy energy calculations; small system validation | Very High |
| Density Functional Theory (DFT) | Uses electron density rather than wave function as fundamental variable | Geometry optimization; transition state searching; medium-sized systems | Moderate |
| Semi-empirical Methods | Simplified quantum mechanics with empirical parameters | Large system screening; conformational analysis | Low |
| Molecular Mechanics | Classical force fields without electronic structure | Very large systems; protein-ligand interactions | Very Low |
Protocol 1: Transition State Optimization
Initial Structure Preparation: Generate reasonable guess structures for reactants, products, and putative transition state using chemical intuition and analogous systems.
Geometry Optimization: Employ computational methods (typically DFT) to locate stationary points on the potential energy surface through iterative solution of the Schrödinger equation.
Frequency Calculation: Perform vibrational analysis to confirm transition state (one imaginary frequency) versus minimum (all real frequencies).
Intrinsic Reaction Coordinate (IRC) Analysis: Follow the reaction path from the transition state forward to products and backward to reactants to confirm the mechanism.
Energy Calculation: Compute accurate electronic energies for all stationary points using high-level quantum chemical methods.
Protocol 2: Reaction Pathway Mapping
Reaction Center Identification: Define the atoms directly involved in bond formation and cleavage using techniques such as reaction template extraction [21].
Coordinate Definition: Establish a reaction coordinate describing the progression from reactants to products.
Potential Energy Surface Scan: Calculate energies for structures at regular intervals along the reaction coordinate.
Mechanistic Template Application: Apply expert-coded mechanistic templates to interpret electron movements in chemically meaningful terms [21].
Kinetic Parameter Extraction: Calculate activation energies and rate constants from energy barriers between stationary points.
Applications in Chemical Reaction Mechanisms Research
Mechanistic Pathway Elucidation
The Schrödinger equation enables researchers to move beyond product identification to detailed understanding of reaction mechanisms at the electronic level. Recent advances in large-scale reaction datasets, such as the mech-USPTO-31K dataset containing chemically reasonable arrow-pushing diagrams validated by synthetic chemists, have created new opportunities for mechanism-based reaction prediction [21]. These developments address the critical need for sophisticated chemical models that explicitly capture underlying reaction mechanisms, including step-by-step sequences of electron movements and reactive intermediates [21].
The process of automated mechanistic pathway labeling involves two key steps: reaction template (RT) extraction and mechanistic template (MT) application [21]. Reaction templates are obtained by identifying changed atoms through comparison of chemical environments before and after reactions, then extending to include π-conjugated systems and mechanistically important special groups [21]. Mechanistic templates then describe the actual electron movements in the form of arrow-pushing diagrams, representing attacking and electron-receiving moieties based on chemistry knowledge [21].
Table 3: Key Research Reagent Solutions in Computational Mechanism Studies
| Research Tool | Function | Application Example |
|---|---|---|
| Reaction Template (RT) Libraries | Encodes reaction transformation patterns as computable rules | Automated identification of reaction centers from experimental data [21] |
| Mechanistic Template (MT) Databases | Expert-coded electron movement patterns for common reaction classes | Distinguishing between SN1 and SN2 mechanisms based on chemical environment [21] |
| Quantum Chemistry Software Packages | Numerical solvers for the electronic Schrödinger equation | Calculating transition state geometries and energies for barrier determination |
| Reaction Mechanism Datasets | Curated collections of validated mechanistic pathways | Training machine learning models for reaction outcome prediction [21] |
| Atom-Mapping Algorithms | Automated identification of atom correspondence between reactants and products | Preparing reaction data for mechanistic analysis [21] |
Data Generation and Analysis Framework
The integration of quantum chemistry calculations with automated mechanism generation creates a powerful framework for high-throughput mechanistic studies. The following diagram illustrates this data generation and analysis pipeline:
Diagram 2: Mechanism Analysis Pipeline
This framework addresses significant challenges in computational reaction mechanism research, including the frequent absence of necessary reagents in recorded reaction data [21]. For approximately 60% of reactions in curated datasets, necessary reagents must be added to complete the mechanistic picture [21]. Additionally, the framework incorporates technical maneuverability to capture important mechanistic elements beyond the immediate reaction center, addressing limitations associated with locality constraints [21].
Implications for Drug Development
The application of Schrödinger equation-based computational methods has transformed early-stage drug discovery by providing atomic-level insights into molecular interactions and reactivity. Quantum chemical calculations enable researchers to predict metabolic pathways, assess potential toxicity, and optimize synthetic routes before undertaking costly experimental work.
In pharmaceutical research, understanding reaction mechanisms is crucial for interpreting product formation at the atomic and electronic level [21]. Computational models that explicitly capture underlying reaction mechanisms provide valuable insights into stereochemistry, reaction kinetics, byproduct formation, and other important reaction details [21]. This mechanistic understanding is particularly valuable for predicting drug metabolism and identifying potential reactive metabolites that could cause toxicity.
The development of reliable reaction outcome prediction models based on mechanistic understanding represents an active research frontier [21]. Such models aim to predict the same arrow-pushing diagrams that human chemists would draw, capturing the finer details of electron movements and reactive intermediates that are crucial for comprehensive reaction understanding [21]. As these models improve, they will increasingly support synthetic planning in drug development by identifying viable synthetic pathways to target compounds [21].
The Schrödinger equation provides the essential theoretical foundation for computational chemistry and its application to reaction mechanism research. By enabling first-principles calculations of molecular structure, properties, and reactivity, this fundamental equation has transformed our ability to understand and predict chemical behavior at the most detailed level. The continuing development of computational methods, coupled with growing mechanistic datasets and more accurate quantum chemical models, promises to further enhance our capability to explore chemical reaction spaces and accelerate drug development processes. As computational power increases and algorithms become more sophisticated, the role of Schrödinger equation-based calculations in pharmaceutical research will continue to expand, ultimately enabling more efficient and predictive drug discovery.
A Toolbox for the Digital Chemist: AI, QM, and Hybrid Methods in Action
The computational exploration of chemical reaction mechanisms represents a cornerstone of modern chemical research, with profound implications for drug development, materials science, and synthetic chemistry. Traditional approaches to reaction prediction have often relied on quantum chemical calculations, which are computationally prohibitive for large-scale exploration, or data-driven models that frequently violate fundamental physical laws. The core challenge has been bridging the gap between predictive accuracy and mechanistic understanding. Recent generative artificial intelligence (AI) breakthroughs, specifically Flow Matching for Electron Redistribution (FlowER) and Equivariant Consistency Models (ECTS), are redefining this landscape. These approaches integrate physical constraints directly into their architectures, enabling not only accurate prediction of reaction outcomes but also providing unprecedented insight into the electron-level pathways that govern chemical reactivity.
FlowER: Electron-Conscious Reaction Prediction
Core Principle and Architecture
FlowER recasts reaction prediction as a problem of electron redistribution within the deep generative framework of flow matching [22] [23]. Its foundational innovation lies in explicitly conserving both mass and electrons through the bond-electron (BE) matrix representation, a concept originally developed by chemist Ivar Ugi in the 1970s [8] [24]. This representation uses a matrix where nonzero values represent bonds or lone electron pairs and zeros represent their absence, providing a direct computational analogue for tracking electron movement during reactions [8].
The model employs flow matching to learn a probability path between the electron distribution of reactants and that of products [22]. This approach conceptually aligns with the "arrow-pushing" formalism taught to chemists, where curved arrows show the movement of electrons during bond formation and breakage [25]. By operating directly on the BE matrix, FlowER inherently respects conservation laws that previous models based on SMILES strings or molecular graphs often violated, eliminating "hallucinatory" failure modes where atoms spontaneously appear or disappear [8] [23].
Detailed Methodology and Workflow
The experimental implementation of FlowER involves a carefully designed pipeline that transforms chemical structures into a flow-matched generative process. The following diagram illustrates the core workflow of the FlowER framework for predicting reaction mechanisms.
Input Representation and Training:
- Input Format: FlowER requires atom-mapped reactions for training, validation, and testing. Each elementary reaction step follows the format:
mapped_reaction|sequence_idx[25]. - Training Data: The model is trained on a combination of subsets from USPTO-FULL, RmechDB, and PmechDB, totaling over a million chemical reactions primarily from U.S. Patent Office databases [25] [8].
- Key Hyperparameters: Critical configuration includes embedding dimension size (
emb_dim), number of transformer layers (enc_num_layers), attention heads (enc_heads), and radial basis function parameters for the BE matrix representation (rbf_low,rbf_high,rbf_gap) [25].
Inference and Search Protocol:
- Beam Search: For mechanistic exploration, FlowER employs beam search to identify plausible reaction pathways. Users input reactions in the format
reactant>>product1|product2|...in a specified text file [25]. - Search Parameters: Key beam search configurations include
beam_size(top-k candidate selection),nbest(cutoff for top-k outcomes),max_depth(maximum exploration depth), andchunk_size(concurrent processing of reactant sets) [25]. - Pathway Visualization: The generated routes can be visualized using the provided
vis_network.ipynbJupyter notebook [25].
Research Reagent Solutions
Table 1: Essential Computational Resources for FlowER Implementation
| Research Reagent | Function | Specifications |
|---|---|---|
| FlowER Codebase | Core model architecture and training pipelines | Available via GitHub [25] |
| Mechanistic Dataset | Training data with elementary reaction steps | Includes USPTO-FULL, RmechDB, PmechDB [25] |
| Computational Environment | Hardware/software infrastructure | Ubuntu ≥16.04, Conda ≥4.0, GPU with 25GB+ Memory, CUDA ≥12.2 [25] |
| Bond-Electron Matrix | Physical representation of molecules | Encodes bonds and lone electron pairs; ensures mass/electron conservation [8] |
ECTS: Ultra-Fast Exploration of Transition States
Core Principle and Architecture
The Equivariant Consistency Model for Transition State (ECTS) represents a complementary breakthrough focused on the critical structures governing reaction kinetics: transition states (TS) [26]. Traditional transition state exploration requires extensive quantum chemistry calculations, making mechanistic studies computationally prohibitive for complex systems. ECTS addresses this bottleneck by unifying TS generation, energy barrier prediction, and reaction pathway mapping within a single, ultra-fast diffusion framework [26].
The model builds upon consistency model principles, which enable direct mapping from noise to data instead of iterative reverse-time differential equation solving [26]. By incorporating equivariant constraints, ECTS respects the geometric symmetries of molecular systems, ensuring generated structures are physically meaningful. This approach achieves an efficiency at least two orders of magnitude higher than conventional diffusion models while maintaining remarkable accuracy [26].
Detailed Methodology and Workflow
ECTS operates through a streamlined diffusion process that directly generates transition state geometries and associated energy barriers. The following diagram illustrates its efficient single-step or few-step denoising process.
Consistency Diffusion Process:
- Mathematical Foundation: ECTS employs a modified probability flow ordinary differential equation (ODE) that transforms the molecular conformation distribution into a tractable noise distribution while maintaining SE(3)-equivariance [26]. This allows direct mapping from noise vectors to transition state structures without iterative reverse-time solving.
- Sampling Efficiency: Where traditional diffusion models require thousands of denoising steps, ECTS achieves comparable quality with only one sampling step, with performance improving further with a few additional iterations [26].
Performance Metrics:
- Structural Accuracy: Generated TS structures exhibit an exceptionally small error margin of just 0.12 Å root mean square deviation compared to ground truth quantum chemical calculations [26].
- Energy Prediction: Through continuous refinement during denoising, ECTS predicts energy barriers with a median error of merely 2.4 kcal/mol without post-DFT calculations [26].
- Pathway Generation: The model generates complete reaction paths that show strong agreement with true reaction coordinates, enabling mechanistic exploration [26].
Research Reagent Solutions
Table 2: Essential Computational Resources for ECTS Implementation
| Research Reagent | Function | Specifications |
|---|---|---|
| ECTS Framework | Transition state generation and pathway exploration | Implements equivariant consistency diffusion [26] |
| Quantum Chemistry Data | Training data with validated transition states | Structures and energies from ab initio calculations [26] |
| Equivariant Networks | SE(3)-equivariant transformer architecture | Encodes Cartesian molecular conformations [26] |
| Consistency Sampler | Ultra-fast sampling from noise to data | Enables single-step generation [26] |
Comparative Analysis and Performance Benchmarks
Quantitative Performance Metrics
Table 3: Performance Comparison of FlowER and ECTS Against Traditional Methods
| Model | Key Innovation | Accuracy Metric | Efficiency Gain | Physical Constraints |
|---|---|---|---|---|
| FlowER | Electron flow matching | Matches/exceeds existing approaches in finding standard mechanistic pathways [22] | Enables rapid pathway search via beam search [25] | Explicit mass and electron conservation [8] |
| ECTS | Consistency diffusion | 0.12 Å RMSD for TS structures; 2.4 kcal/mol median error for barriers [26] | 100x faster than conventional diffusion models [26] | SE(3)-equivariance for geometrically valid structures [26] |
| Traditional AI Models | SMILES-based or graph-based | Limited by hallucinatory failure modes [8] | Varies by approach | Often violate conservation laws [8] |
| Quantum Chemistry | First-principles calculations | Ground truth but computationally limited | Computationally prohibitive for large systems | Physically rigorous but resource-intensive |
Integration Potential for Reaction Discovery
The complementary strengths of FlowER and ECTS suggest powerful integration potential. FlowER provides the electron-level understanding of reaction sequences, while ECTS delivers ultra-fast transition state characterization with kinetic parameters. Together, they form a comprehensive framework for computational reaction exploration:
- Mechanism Elucidation: FlowER can propose plausible electron redistribution pathways, which ECTS can then validate through transition state analysis [22] [26].
- Kinetic Profiling: The energy barriers predicted by ECTS provide critical information about reaction feasibility and rates that complement FlowER's mechanistic insights [26].
- Pathway Optimization: Researchers can use these tools iteratively to explore reaction networks, identify rate-limiting steps, and design improved synthetic routes.
Implementation Guide for Research Applications
Practical Deployment Considerations
For FlowER Implementation:
- Environment Setup: Begin with the GitHub repository
FongMunHong/FlowERand follow the system requirements (Ubuntu ≥16.04, Conda ≥4.0, GPU with 25GB+ memory, CUDA ≥12.2) [25]. - Data Preparation: Structure input data according to the required format for elementary reaction steps (
mapped_reaction|sequence_idx) [25]. - Beam Search Configuration: For reaction exploration, modify
beam_size,nbest, andmax_depthparameters insettings.pyto balance comprehensiveness and computational cost [25].
For ECTS Application:
- Transition State Generation: Input molecular graphs to generate candidate transition state structures with associated energy barriers [26].
- Pathway Mapping: Use the generated reaction paths to visualize complete reaction coordinates and identify key intermediates [26].
- Validation: Despite the high accuracy, critical reactions should be validated with targeted quantum chemical calculations [26].
Current Limitations and Development Trajectory
Both technologies represent rapidly evolving frontiers with identifiable growth paths:
FlowER Limitations:
- Chemistry Coverage: The current model has limited exposure to reactions involving certain metals and catalytic cycles, as these are underrepresented in its training data [8] [24].
- Scalability: While efficient for single-step reactions, multi-step pathway prediction remains computationally intensive [25].
ECTS Limitations:
- System Complexity: Performance on large, flexible molecular systems with multiple rotatable bonds requires further validation [26].
- Accuracy Boundaries: While impressive, the 2.4 kcal/mol median error may still be significant for reactions with very small energy differences [26].
Development Roadmap:
- Near-term: Expansion to organometallic and catalytic reactions, improved handling of complex reaction networks [8].
- Long-term: Full integration with automated synthesis planning and experimental validation platforms [8] [24].
FlowER and ECTS represent paradigm shifts in the computational exploration of chemical reaction mechanisms. By embedding physical principles directly into generative AI architectures—electron conservation through BE matrices in FlowER and geometric equivariance in ECTS—these approaches overcome fundamental limitations of previous data-driven models. They provide researchers with unprecedented capabilities to map reaction pathways, predict products with high accuracy, characterize transition states, and estimate kinetic parameters at computational speeds previously unimaginable.
For the drug development professionals and research scientists comprising the target audience, these tools offer practical solutions for accelerating reaction discovery and optimization. The open-source nature of FlowER ensures accessibility, while the demonstrated performance of both technologies provides confidence in their application to challenging problems in synthetic chemistry, medicinal chemistry, and materials science. As these frameworks continue to evolve and integrate, they promise to significantly advance our fundamental understanding of chemical reactivity while dramatically accelerating the design and discovery of new molecules with tailored properties.
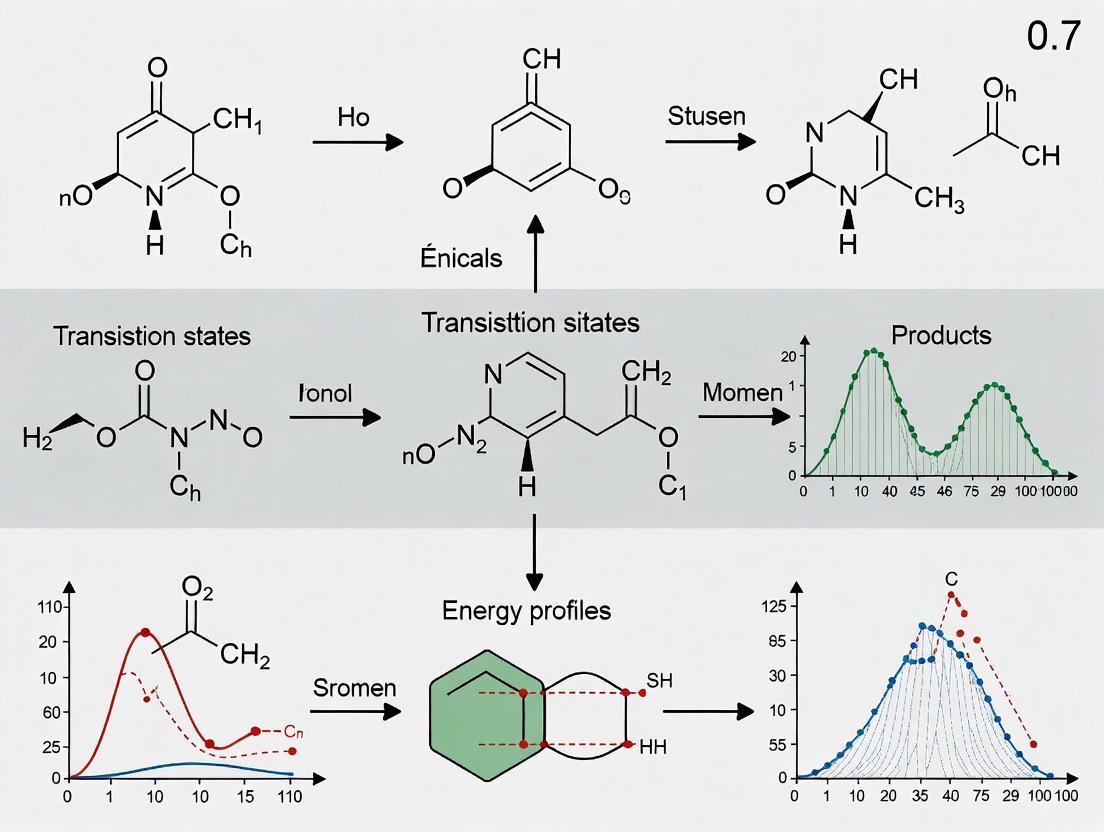
The computational exploration of chemical reaction mechanisms is a cornerstone of modern chemistry, crucial for advancing catalyst design, understanding reaction kinetics, and accelerating drug development. Traditional methods for mapping Potential Energy Surfaces (PES) to identify intermediates and transition states are often hampered by exponential pathway complexity and substantial computational costs. The integration of Large Language Models (LLMs) into specialized computational programs is emerging as a transformative solution to these challenges. This whitepaper examines the pioneering role of LLMs in guiding automated reaction pathway exploration, with a focused analysis on the ARplorer program. We detail its architecture, the LLM-guided chemical logic that powers its efficiency, and the experimental protocols that enable its application in studying complex multi-step reactions.
ARplorer is an automated computational program, built using Python and Fortran, designed to conduct fast and efficient exploration of reaction pathways for PES studies [4]. Its development addresses a critical limitation in conventional quantum mechanics (QM) and molecular dynamics (MD) approaches: the absence of chemical logic implementation based on existing literature and the need for system-specific modifications [4].
The program operates on a recursive algorithm, with each iteration involving three core steps [4]:
- Active Site Identification: The program identifies active sites and potential bond-breaking locations to set up multiple input molecular structures for analysis.
- Transition State Search & Optimization: It employs a blend of active-learning sampling and potential energy assessments to optimize molecular structures and hone in on potential intermediates.
- Pathway Validation via IRC: Intrinsic Reaction Coordinate (IRC) analysis is performed to derive new reaction pathways from optimized structures, eliminate duplicates, and finalize structures for subsequent input [4].
For efficiency, ARplorer combines faster semi-empirical methods (GFN2-xTB) for PES generation with more precise algorithms (e.g., from Gaussian 09) for TS searches, though it maintains the flexibility to switch to Density Functional Theory (DFT) for higher precision when necessary [4].
Workflow Visualization
The following diagram illustrates the core recursive workflow of the ARplorer program:
The Core Innovation: LLM-Guided Chemical Logic
The pivotal innovation within ARplorer is its LLM-guided chemical logic, which moves beyond unfiltered PES searches by applying predetermined, chemically plausible biases to refine the search process [4]. This logic is built from two complementary knowledge sources, creating a powerful hybrid system for pathway prediction.
The Scientist's Toolkit: Key Research Reagent Solutions
| Component | Function & Explanation |
|---|---|
| General Chemical Knowledge Base | A curated collection of indexed data from textbooks, databases, and research articles. Serves as the foundational source of established chemical rules for the LLM [4]. |
| Specialized LLM | A fine-tuned large language model that processes the general knowledge base and system-specific SMILES strings to generate targeted chemical logic and reaction patterns [4]. |
| SMILES Strings | Simplified Molecular-Input Line-Entry System; a textual representation of the molecular structure. Serves as the primary input for generating system-specific chemical logic [4]. |
| SMARTS Patterns | A powerful extension of SMILES that describes molecular patterns and functional groups. Used by the LLM to encode reaction rules for identifying plausible reaction sites [4]. |
| Python Pybel Module | A Python module used to compile lists of active atom pairs and potential bond-breaking locations based on the generated chemical logic [4]. |
| GFN2-xTB | A semi-empirical quantum mechanical method. Used for quick generation of potential energy surfaces and large-scale screening within the ARplorer workflow [4]. |
| DFT (e.g., Gaussian 09) | Density Functional Theory software. Used for more precise and detailed quantum mechanical calculations when higher accuracy is required [4]. |
Constructing the Chemical Logic Library
The process of building the chemical logic library is methodical and occurs prior to the autonomous PES exploration. It ensures that ARplorer's searches are grounded in established chemical knowledge.
- General Chemical Logic Generation: The process begins with pre-screened data sources (books, databases, articles) being processed and indexed to form a general chemical knowledge base. This base is refined using prompt engineering to generate general SMARTS patterns, which are formalized representations of chemical rules [4].
- System-Specific Chemical Logic Generation: For a given reaction system, the reactants are converted into SMILES format. This textual representation, along with prompts designed to access the general knowledge base, is fed into a specialized LLM. The LLM then generates targeted chemical logic and SMARTS patterns specific to the system under investigation [4].
It is critical to emphasize that in the current ARplorer workflow, the LLM serves exclusively as a literature mining tool during this initial knowledge curation phase. The model is not involved in energy evaluation or pathway ranking. All assessments of reaction plausibility and kinetics are performed exclusively via first-principles QM computations, guaranteeing the quantum chemical rigor of the results [4].
Knowledge Curation Visualization
The following diagram illustrates the process of building the chemical logic library:
Experimental Protocols & Performance Benchmarks
The integration of LLM-guided chemical logic with active-learning TS sampling and parallel computing creates a highly efficient workflow for mechanistic investigation. The following protocol outlines a typical application of ARplorer for studying a multi-step reaction.
Detailed Experimental Protocol for Multi-Step Pathway Exploration
- System Preparation & Logic Curation
- Input: Provide the SMILES strings of the reactant molecules.
- Action: Execute the knowledge curation workflow (Section 3.1) to generate the system-specific chemical logic and SMARTS patterns. This logic is stored in a library for the exploration phase.
- Initialization
- Input: Load the initial reactant geometry and the curated chemical logic library.
- Action: The program uses Pybel to identify initial active atom pairs and potential bond-breaking locations based on the chemical logic [4].
- Recursive Pathway Exploration
- For each intermediate generated: a. Structure Setup: Multiple input structures are configured based on the identified active sites. b. TS Search & Optimization: An iterative TS search is launched using a hybrid GFN2-xTB/Gaussian 09 approach. The active-learning method efficiently hones in on viable transition states [4]. c. IRC & Validation: For each optimized TS structure, an IRC calculation is performed in both directions to confirm it connects to the expected reactants and products. New intermediates and products are added to the exploration queue [4]. d. Filtering & Duplicate Removal: Energy filters and duplicate checks are applied. Pathways deemed implausible by the chemical logic or with high-energy barriers are discarded.
- Termination & Output
- The process terminates when no new viable pathways are found within a specified energy window.
- Output: The program returns a comprehensive list of discovered reaction pathways, including all intermediates, transition states, and their associated thermodynamic and kinetic parameters.
Quantitative Performance Data
The effectiveness of ARplorer has been demonstrated through case studies on complex multi-step reactions, including organic cycloadditions, asymmetric Mannich-type reactions, and organometallic Pt-catalyzed reactions [4]. The table below summarizes the key performance enhancements offered by its integrated approach.
Table 1: Performance Advantages of the ARplorer Framework
| Feature | Benefit & Quantitative Impact |
|---|---|
| LLM-Guided Chemical Logic | Applies literature-derived and system-specific chemical rules to filter out implausible pathways, drastically reducing the search space and computational cost compared to unfiltered searches [4]. |
| Active-Learning TS Sampling | Enhances the efficiency and speed of transition state localization, a traditionally time-consuming step in QM calculations [4]. |
| Energy Filter-Assisted Parallel Computing | Minimizes unnecessary computations by running multiple reaction searches in parallel and filtering them based on energetic criteria [4]. |
| Multi-Step Reaction Searches | Demonstrates versatility and effectiveness in automating the exploration of complex, multi-step reaction pathways, as shown in case studies [4]. |
The paradigm of using LLMs for molecular reasoning is also being advanced by other research. For instance, the atom-anchored LLM framework demonstrates how general-purpose LLMs can be guided to perform precise chemical tasks like retrosynthesis without task-specific training [27]. This approach, which anchors chain-of-thought reasoning to unique atomic identifiers in a SMILES string, has achieved high success rates in identifying chemically plausible disconnection sites (≥90%) and final reactants (≥74%) [27].
The integration of Large Language Models into tools like ARplorer marks a significant leap forward in the computational exploration of chemical reaction mechanisms. By moving beyond a purely data-driven paradigm to one augmented by curated chemical knowledge, these systems achieve a new level of efficiency and practicality. ARplorer exemplifies this synergy, combining the physical rigor of quantum mechanics with the pattern-recognition and knowledge-synthesis capabilities of LLMs. This powerful combination allows researchers to tackle increasingly complex organic and organometallic systems, accelerating the discovery of new reactions and catalysts. As LLM technology continues to evolve, its role as an integral component in the computational chemist's toolkit is set to expand, further bridging the gap between theoretical exploration and practical reaction development.
The computational exploration of chemical reaction mechanisms presents a fundamental challenge: the reactions of interest occur at the quantum mechanical level, while they are profoundly influenced by their molecular environment, which may encompass thousands to millions of atoms. No single computational method can simultaneously provide electronic-level accuracy and simulate biologically or chemically relevant time and length scales. This challenge has given rise to the multiscale modeling paradigm, which strategically combines different levels of theory to bridge these scales. By integrating quantum mechanics (QM), molecular mechanics (MM), molecular dynamics (MD), and, more recently, machine learning potentials (MLPs), researchers can now simulate chemical reactions with unprecedented accuracy and scope. These methods are particularly vital in fields like drug design, where understanding reaction pathways and binding events in atomic detail is essential but occurs within massively complex cellular environments [28].
The core strength of multiscale modeling lies in its targeted application of computational resources. QM methods, though accurate, are prohibitively expensive for large systems. MM methods, which use classical force fields, can handle large systems but cannot model bond breaking and formation. Multiscale methods overcome these limitations by partitioning the system, applying a high-level theory like QM only to the chemically active region (e.g., an enzyme's active site), and treating the vast surroundings with a computationally efficient MM potential [29]. The emergence of MLPs, trained on QM data, now offers a third pathway, promising near-QM accuracy at a fraction of the computational cost, thereby accelerating the exploration of reaction mechanisms and free energy landscapes [30] [31].
Foundational Methodologies in Multiscale Modeling
The Quantum Mechanics/Molecular Mechanics (QM/MM) Framework
The QM/MM approach, for which the 2013 Nobel Prize in Chemistry was awarded, is a cornerstone of multiscale modeling. It seamlessly integrates a QM description for the region where the chemistry occurs (e.g., a reacting substrate and key catalytic residues) with an MM description for the surrounding environment (e.g., protein scaffold, solvent water) [28] [29]. The total energy of the system is expressed as:
[ E{\text{total}} = E{\text{QM}} + E{\text{MM}} + E{\text{QM-MM}} ]
Here, ( E{\text{QM}} ) is the energy of the quantum region, ( E{\text{MM}} ) is the energy of the classical region, and ( E_{\text{QM-MM}} ) is the interaction term that couples the two regions. This coupling is a critical aspect of the model and is typically handled through one of several embedding schemes:
- Mechanical Embedding: The simplest scheme, where the QM-MM interactions are treated entirely with the classical MM force field.
- Electrostatic Embedding: A more sophisticated and common approach where the MM point charges are included in the Hamiltonian for the QM calculation. This allows the quantum region to be polarized by the classical environment, a crucial effect for accurate modeling of reactions in polar solvents or proteins [31] [29].
QM/MM has proven indispensable for studying enzyme catalysis, revealing cryptic drug binding sites, and predicting drug resistance mechanisms by providing a "computational microscope" on biological processes [28].
Molecular Dynamics (MD) for Sampling and Dynamics
Molecular Dynamics simulations complement QM/MM by providing the means to simulate the time-dependent evolution of a system. While MM-based MD is a workhorse for studying conformational changes and dynamics of large biomolecules, it cannot simulate electronic processes. In a multiscale context, MD is used to sample the configurations of the MM environment, which in turn affects the QM region. Furthermore, ab initio MD, where forces are computed on-the-fly from QM calculations, can be combined with MM in QM/MM-MD simulations to model reactive processes. The ability of MD to access microsecond to millisecond timescales for large systems makes it a powerful tool for understanding the dynamic nature of drug targets and the complexity of biological systems [28].
The Rise of Machine Learning Potentials (MLPs)
Machine Learning Interatomic Potentials represent a paradigm shift. MLPs are trained on high-quality QM data to predict potential energies and atomic forces, achieving near-QM accuracy while maintaining a computational cost comparable to classical MM force fields [31]. This breakthrough addresses the primary bottleneck of QM/MM methods: the high computational expense of the QM calculation. MLPs can be integrated into multiscale simulations in two primary ways:
- As a direct replacement for the QM region in a new hybrid scheme known as ML/MM (Machine Learning/Molecular Mechanics) [30] [31].
- As a standalone force field for large-scale simulations of materials and molecules, enabling the study of complex transformations, such as the structural reconstruction of nanodiamonds, with DFT-level accuracy [32].
Table 1: Comparison of Core Computational Methods
| Method | Fundamental Principle | Typical System Size | Key Strengths | Primary Limitations |
|---|---|---|---|---|
| Quantum Mechanics (QM) | Solves electronic Schrödinger equation | 10s - 100s of atoms | High accuracy; models bond breaking/formation | Extremely high computational cost |
| Molecular Mechanics (MM) | Classical balls-and-springs force field | 100,000s - millions of atoms | Fast; excellent for large systems | Cannot model reactions; accuracy depends on parameterization |
| QM/MM | Combines QM and MM via an interface | Entire proteins in solvent | Models chemistry in realistic environments; includes polarization | Computational cost dominated by QM region |
| ML/MM | Replaces QM with a ML potential in a hybrid scheme | Entire proteins in solvent | Near-QM accuracy at MM cost; excellent for sampling | Requires high-quality QM training data; transferability concerns |
Advanced Hybrid Schemes: ML/MM and Beyond
The development of ML/MM is a natural evolution of the QM/MM paradigm. By substituting the computationally expensive QM calculation with a fast and accurate MLP, ML/MM dramatically expands the scope of problems that can be addressed [30]. The total energy expression is analogous to QM/MM:
[ E{\text{total}} = E{\text{ML}} + E{\text{MM}} + E{\text{ML-MM}} ]
where ( E_{\text{ML}} ) is the energy from the machine learning potential. Several strategies have been developed to couple the ML and MM regions effectively [30]:
- Mechanical Embedding (ME): The ML region interacts with the fixed atomic charges of the MM region via classical electrostatic and Lennard-Jones potentials. This is efficient and widely supported but does not allow polarization of the ML region by the MM environment [31].
- Polarization-Corrected Mechanical Embedding (PCME): A vacuum-trained ML potential is supplemented with post-hoc electrostatic corrections to approximate environmental effects, preserving model transferability.
- Environment-Integrated Embedding (EIE): The ML potential is trained with explicit inclusion of MM-derived electrostatic fields, leading to higher accuracy but requiring specialized training data.
The application of ML/MM is particularly impactful in free energy calculations, which are crucial for predicting binding affinities in drug design. A recent implementation in the AMBER simulation package introduced a novel thermodynamic integration (TI) framework compatible with ML/MM. This framework achieved an accuracy of 1.0 kcal/mol for hydration free energies, outperforming traditional approaches. By enabling more precise conformational sampling, ML/MM provides a new avenue for reliable free energy predictions, a cornerstone of rational drug development [31].
Practical Implementation and Workflows
The Scientist's Toolkit: Essential Software and Reagents
Table 2: Key Research Reagent Solutions for Multiscale Modeling
| Item Name / Software | Category | Primary Function in Multiscale Modeling |
|---|---|---|
| ORCA | Quantum Chemistry Software | Performs QM, QM/MM, QM1/QM2, and QM1/QM2/MM calculations; geometry optimization and transition state search. |
| AMBER | Molecular Dynamics Suite | Facilitates classical MD, ML/MM simulations, and advanced free energy calculation methods (TI, FEP). |
| ANI-2x | Machine Learning Potential | A neural network potential providing near-DFT accuracy for organic molecules; used in ML/MM for the ML region. |
| CellPack | Mesoscale Modeling Tool | Models complex biomolecular systems at the mesoscale, enabling integration from atomic to cellular scales. |
| pdbtoORCA | Workflow Automation | A Python code for setting up multiscale calculations in ORCA, defining QM and MM regions from PDB files. |
A Standard Workflow for SN2 Reaction Analysis
The following diagram illustrates a generalized workflow for studying a reaction mechanism, such as an SN2 reaction, using multiscale methods. This workflow synthesizes the steps involved in studies like those cited, which used multiscale methods to accurately reproduce transition state geometries and energetics [29].
Detailed Protocol: Setting Up a QM/MM Calculation in ORCA
This protocol is adapted from a study assessing multiscale methods for SN2 reactions and Claisen rearrangements [29].
System Preparation:
- Obtain the initial atomic coordinates of the entire system (e.g., the solute and explicit solvent molecules) in a PDB file format.
- For the studied SN2 reaction (CH₃I + NH₂OH/NH₂O⁻), the system would include the reacting molecules solvated in a box of water molecules.
Region Definition in the PDB File:
- QM Region: Specify the atoms to be treated quantum mechanically by setting the occupancy column to
1.00in the PDB file. For the SN2 reaction, this would include the carbon and iodine of methyl iodide and the nitrogen and oxygen of the nucleophile. - Active MM Region: Define the atoms that will be allowed to move during geometry optimization by setting the B-factor column to
1.00. This typically includes the QM region atoms plus a surrounding shell of solvent molecules. - MM Fixed Region (Extension Shell): This region consists of atoms outside the active region that are included in the calculation but have their positions constrained to ensure stable optimization. ORCA can automatically determine this shell based on distance or covalent bonding to the active atoms.
- QM Region: Specify the atoms to be treated quantum mechanically by setting the occupancy column to
ORCA Input File Configuration:
- An example input block for a QM/MM calculation in ORCA is provided below. This specifies the method, basis set, and the treatment of the different regions.
In this example, the
Optkeyword triggers a geometry optimization, which will be performed only on the active atoms.
- An example input block for a QM/MM calculation in ORCA is provided below. This specifies the method, basis set, and the treatment of the different regions.
In this example, the
Execution and Analysis:
- Run the ORCA calculation and analyze the output files for optimized geometries, energies, and atomic forces.
- Compare the calculated activation energy (the energy difference between the reactant and transition state) with experimental data to validate the method.
Applications and Case Studies in Reaction Mechanism Research
Elucidating Solvent Effects on SN2 Reactions and Claisen Rearrangement
Multiscale methods excel at capturing explicit solvent effects that continuum models often miss. A 2024 study systematically applied QM/MM and related methods to two classic reactions [29]:
- SN2 Reactions: The research investigated the reactions of methyl iodide with NH₂OH and NH₂O⁻. The results underscored that explicitly including solvent molecules in the MM region was critical to accurately reproducing the transition state geometry and energetics. The calculated activation energies from multiscale methods showed promising agreement with expected values, unlike calculations in vacuum. The study also highlighted that the size of the active MM region significantly impacts the accuracy of the results.
- Claisen Rearrangement: For the rearrangement of 8-(vinyloxy)dec-9-enoate, both QM-only and multiscale methods correctly identified the reaction pathway. However, activation free energies calculated using a continuum solvation model were less accurate. Multiscale methods that explicitly included solvent molecules in the MM region more effectively captured the solvent's influence on the activation barrier.
Accelerated Free Energy Calculations with ML/MM
A groundbreaking application of ML/MM is its use in rigorous free energy calculations. A 2025 study developed a hybrid ML/MM interface within the AMBER package and created a new thermodynamic integration (TI) framework to overcome the challenge of applying MLPs in TI calculations [31]. The key innovation was a revised TI scheme that perturbs only the non-bonded interactions between the ML and MM regions, introducing a "reorganization energy" term to compensate for the lack of perturbation within the ML region itself. This approach allowed the calculation of hydration free energies with an accuracy of 1.0 kcal/mol, outperforming traditional methods. This demonstrates the power of ML/MM to provide a more accurate and efficient foundation for predicting binding affinities, a critical task in drug discovery.
Modeling Complex Structural Transformations in Materials
Beyond molecular chemistry, MLP-accelerated multiscale simulations are making an impact in materials science. In a study of nanodiamond (ND) structural reconstruction, a machine learning potential was developed to simulate NDs comprising thousands of atoms over nanosecond timescales with DFT accuracy [32]. The simulations revealed a complex, multistage transformation pathway involving graphitization, atomic migration, and a self-healing process. This case study highlights the power of MLPs to provide atomistic insight into complex, multiscale structural transformations that are otherwise inaccessible to direct QM simulation.
The multiscale modeling arsenal, encompassing QM/MM, MD, and MLPs, provides a powerful and flexible framework for the computational exploration of chemical reaction mechanisms. By strategically combining the accuracy of quantum mechanics with the scalability of classical force fields and the speed of machine learning, researchers can now tackle problems that were once computationally intractable. As highlighted throughout this guide, these methods are indispensable for modeling reactions in realistic environments, such as solvents and enzymes, and for achieving highly accurate predictions of key properties like binding free energies.
The future of multiscale modeling is being shaped by the convergence of improved algorithms, powerful computing architectures, and the growth of rich, diverse datasets [28]. The emergence of ML/MM is a clear sign of this evolution, blending physical chemistry with modern data science [30]. As these tools become more integrated, automated, and accessible through platforms like ORCA and AMBER, their impact will grow. The ongoing development of highly accurate, data-rich, physics-based multiscale approaches is poised to realize its long-promised impact, driving forward the discovery and design of novel therapeutics and materials through an ever-deepening understanding of chemical complexity from the atom to the cell.
The processes of lead optimization and catalyst design represent two of the most formidable bottlenecks in traditional pharmaceutical research and development. Lead optimization, the iterative process of transforming a promising "hit" compound into a viable drug candidate with favorable pharmacokinetic and safety properties, has conventionally required synthesizing and testing thousands of analogs over several years, with high associated costs [33]. Similarly, predicting and optimizing catalytic reactions for synthetic efficiency has relied heavily on experimental trial-and-error. However, the integration of artificial intelligence (AI) and advanced computational modeling is fundamentally rewiring these workflows, shifting the center of gravity from the wet lab to the computer—from in vitro to in silico [34]. This paradigm shift enables a "predict-then-make" approach, where hypotheses are generated, molecules are designed, and properties are validated computationally at a massive scale, with precious laboratory resources reserved for confirming the most promising, AI-vetted candidates [35] [34].
Framed within the broader computational exploration of chemical reaction mechanisms, these advancements are not merely about accelerating existing processes but about enabling entirely new capabilities. Modern generative AI models can now propose novel molecular structures with optimized properties and predict complex reaction outcomes while adhering to fundamental physical constraints like the conservation of mass and energy [8] [26]. This technical guide examines the core AI technologies driving this change, provides detailed methodologies for their implementation, and quantifies their tangible impact on the speed and success of pharmaceutical R&D.
AI-Driven Transformation of Lead Optimization
Core Technologies and Workflows
The lead optimization phase is being revolutionized by a suite of AI technologies that compress the traditional design-make-test-analyze (DMTA) cycle. Key computational approaches include:
- Generative AI for Molecular Design: Models such as generative adversarial networks (GANs) and diffusion models can design de novo molecular structures optimized for multiple parameters simultaneously, including potency, selectivity, and pharmacokinetic properties [36] [35]. These models explore the chemical space more efficiently than human-guided design, generating novel scaffolds that might otherwise be overlooked.
- Predictive ADMET Panels: Machine learning models, particularly supervised learning, are trained on vast datasets of chemical structures and associated biological assay results to predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles in silico [37] [35]. These panels serve as computational analogues to standard experimental assays, allowing for early assessment of off-target liabilities and other key properties.
- Ultra-Large-Scale Virtual Screening: Instead of physically testing every compound, AI algorithms can evaluate millions of chemical structures for likely activity against a target, dramatically narrowing down the number of compounds that require synthesis and testing [35] [38].
The integration of these technologies creates a powerful, iterative workflow. The following diagram illustrates this closed-loop, AI-accelerated lead optimization system:
Quantitative Impact and Real-World Efficacy
The implementation of AI-driven lead optimization is yielding measurable reductions in both time and cost. The following table summarizes key performance metrics from real-world applications and industry reports:
Table 1: Quantitative Impact of AI on Lead Optimization Metrics
| Metric | Traditional Approach | AI-Accelerated Approach | Example / Source |
|---|---|---|---|
| Time to Clinical Candidate | ~4-6 years [35] | 12-18 months [35] [39] | Exscientia's DSP-1181; Insilico Medicine's IPF drug [35] [39] |
| Compounds Synthesized | Thousands [39] | 10x fewer compounds [39] | Exscientia's CDK7 inhibitor program (136 compounds) [39] |
| Phase I Success Rate | ~40-65% historically [35] | ~85-88% (early data) [35] | 21 of 24 AI-designed molecules passed Phase I [35] |
| Hit Enrichment Rate | Baseline | >50-fold improvement [38] | AI integrating pharmacophoric & protein-ligand data [38] |
A landmark case study involves the company Exscientia, which used an AI-driven platform to design and optimize the compound DSP-1181 for obsessive-compulsive disorder. The project advanced from initiation to clinical trial in just 12 months, a process that traditionally takes about five years. The AI system achieved this by requiring the synthesis and testing of only 350 compounds, compared to an industry standard of approximately 2,500 compounds [35] [39]. This represents an 85% reduction in the number of compounds needed, directly translating to significant time and cost savings.
Computational Advances in Reaction Mechanism Exploration and Catalyst Design
Grounding Predictions in Physical Principles
A significant challenge in applying AI to chemical reaction prediction has been ensuring that the outputs are not just statistically plausible but also physically realistic. Early attempts using large language models (LLMs) often failed to conserve mass or energy, leading to "alchemical" results [8]. Recent research has focused on grounding models in fundamental physical principles.
The FlowER (Flow matching for Electron Redistribution) system, developed at MIT, addresses this by using a bond-electron matrix to represent the electrons in a reaction [8]. This approach, based on a method from chemist Ivar Ugi from the 1970s, uses a matrix with nonzero values to represent bonds or lone electron pairs and zeros to represent their absence. This explicit representation helps conserve both atoms and electrons throughout the reaction prediction process, ensuring outputs adhere to the laws of conservation of mass [8].
Concurrently, ultra-fast diffusion models like ECTS (Equivariant Consistency Generative Model) are unifying transition state (TS) generation, energy prediction, and reaction pathway search within a single framework [26]. ECTS reports an efficiency at least two orders of magnitude higher than conventional diffusion models, with generated TS structures exhibiting an error margin of just 0.12 Å root mean square deviation compared to ground truth and a median energy barrier error of merely 2.4 kcal/mol without post-DFT calculations [26].
Workflow for Computational Reaction Exploration
The following diagram outlines a modern, integrated workflow for exploring chemical reaction mechanisms using these advanced AI tools:
Detailed Methodology: Implementing the FlowER System
For researchers aiming to implement a physically grounded reaction prediction system, the following protocol for the FlowER framework provides a detailed guide:
Objective: To predict the products and detailed mechanism of a chemical reaction while conserving mass and electrons.
Step-by-Step Procedure:
Input Representation:
- Represent the reactant molecules using a bond-electron matrix,
M_reactants. In this matrix, rows and columns correspond to atoms, and matrix elements represent bond orders (e.g., 1 for single, 2 for double) and lone pairs [8]. - This matrix explicitly encodes the electron count for the system.
- Represent the reactant molecules using a bond-electron matrix,
Model Application:
- Input
M_reactantsinto the pre-trained FlowER model. The model employs a flow-matching technique to learn a continuous path for electron redistribution from reactants to products [8]. - The model generates a corresponding product bond-electron matrix,
M_products, and can also infer the intermediate steps of the mechanism.
- Input
Physical Constraint Enforcement:
- The architecture of the model and its training on the bond-electron representation inherently enforce conservation laws. The sum of bond orders and lone pairs (and thus electrons) is conserved between
M_reactantsandM_products[8]. - This step is automatic and does not require post-hoc correction.
- The architecture of the model and its training on the bond-electron representation inherently enforce conservation laws. The sum of bond orders and lone pairs (and thus electrons) is conserved between
Output and Interpretation:
- Decode
M_productsto generate the molecular structures of the reaction products. - Analyze the predicted electron redistribution pathway to infer the detailed reaction mechanism, including the formation and breaking of bonds.
- Decode
Key Resources:
- Source Code and Data: The FlowER model, along with a dataset of mechanistic steps for known reactions, is available open-source on GitHub [8].
- Training Data: The model was trained on over a million chemical reactions from a U.S. Patent Office database [8].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
The effective implementation of the described methodologies relies on a combination of computational tools and data resources. The following table details key solutions essential for modern computational R&D.
Table 2: Key Research Reagent Solutions for Computational R&D
| Tool / Solution | Type | Primary Function in Research |
|---|---|---|
| FlowER Model [8] | Open-Source AI Model | Predicts reaction outcomes and mechanisms while conserving mass and electrons, moving beyond "alchemical" predictions. |
| ECTS Model [26] | Generative AI Model | Unifies transition state generation, energy prediction, and reaction pathway search with high speed and accuracy. |
| In Silico ADMET Panels [37] | Predictive Computational Assay | Provides early assessment of off-target liabilities, toxicity, and key physicochemical properties before synthesis. |
| Generative Molecular Design Platform (e.g., Exscientia's) [39] | Commercial AI Platform | Uses deep learning to propose novel molecular structures that satisfy multi-parameter target product profiles. |
| CETSA (Cellular Thermal Shift Assay) [38] | Experimental Validation Assay | Quantitatively validates direct drug-target engagement in intact cells and tissues, bridging in silico predictions and cellular efficacy. |
| Research Data Products [40] | Curated Data Asset | Provides high-quality, well-governed data (e.g., standardized RNA-seq, integrated EMR/genomics) built on FAIR principles for training robust AI models. |
Integrated Implementation and Future Outlook
The "Lab of the Future": An Integrated Framework
The full potential of AI in lead optimization and catalyst design is realized when computational and experimental workflows are seamlessly integrated into a "lab of the future." This environment is characterized by high levels of automation, connected instruments, and well-integrated data systems where insights from physical experiments and in silico simulations inform each other in real time [40]. According to a Deloitte survey, 53% of biopharma R&D executives reported increased laboratory throughput, and 45% saw a reduction in human error as a direct result of such lab modernization efforts [40].
A mature, predictive lab environment leverages a closed-loop "lab-in-the-loop" platform, where AI proposes new compound variants or experiments, robotics execute the synthesis and testing, and the resulting data is automatically fed back to refine the AI models [36] [35]. This cycle dramatically accelerates the DMTA process. For instance, Genentech's AI-driven "lab-in-the-loop" platform has significantly cut down discovery and lead optimization timelines by reducing the number of cycles needed to arrive at an optimized candidate [35].
Emerging Trends and Challenges
The field continues to evolve rapidly, with several key trends and challenges shaping its trajectory:
- Emerging Trends: The application of Large Language Model (LLM) agents to automate and simulate entire drug discovery workflows is gaining traction [36]. Furthermore, the rise of "self-driving labs" incorporates robotics for automated experimentation under AI-based decision systems to speed up discovery with less human intervention [36]. The market for AI-based pharmaceutical R&D services in North America alone accounted for approximately 50% of the global revenue share in 2024, with the Asia-Pacific region emerging as the fastest-growing market [36].
- Persisting Challenges: Despite progress, significant hurdles remain. A primary challenge is the need for model transparency and explainability, as understanding the rationale behind an AI's prediction is often crucial for scientific validation and regulatory approval [36] [39]. Data governance and sharing restrictions under regulations like GDPR also pose limitations [36]. Finally, while excellent progress has been made with many organic reactions, accurately predicting reactions involving metals and complex catalytic cycles remains an area for future development and model expansion [8].
In conclusion, the integration of advanced computational methods like generative AI and physically grounded reaction predictors is delivering a tangible, positive impact on the speed and efficiency of pharmaceutical R&D. By adopting the detailed methodologies and integrated frameworks outlined in this guide, researchers and drug development professionals are poised to further accelerate the design of high-quality drug candidates and the catalytic reactions to synthesize them, ultimately bringing life-saving medicines to patients more rapidly.
Navigating Computational Limits: Strategies for Enhanced Efficiency and Accuracy
The computational exploration of chemical reaction mechanisms stands as a cornerstone in accelerating drug discovery and materials science. However, this promising field faces a significant constraint: the scarcity of high-quality, labeled experimental data for specific reaction types, which severely limits the application of data-hungry machine learning models. This data scarcity problem is particularly pronounced for novel reaction development and understudied reaction mechanisms where prior knowledge is minimal.
Two complementary machine learning paradigms have emerged as powerful solutions to this challenge. Transfer learning leverages knowledge from data-rich chemical domains (source tasks) to improve performance in data-scarce domains (target tasks) [41] [42]. Active learning strategically selects the most informative experiments to perform, maximizing knowledge gain while minimizing experimental cost [43]. When integrated within a coherent framework, these approaches enable researchers to navigate complex chemical reaction spaces efficiently, even with severely limited starting data.
This technical guide examines the theoretical foundations, practical implementations, and recent advancements in combining active and transfer learning for chemical reaction prediction and optimization, with a specific focus on scenarios characterized by limited reaction types and data availability.
Theoretical Foundations
The Data Scarcity Problem in Chemical Reaction Modeling
Chemical reaction data scarcity manifests in several forms that impact model development:
- Limited reaction examples: Specialized reaction types like Baeyer-Villiger oxidations may have only hundreds of validated examples, insufficient for training complex deep learning models [44].
- Sparse condition sampling: Even for well-studied reactions like Pd-catalyzed cross-couplings, high-throughput experimentation (HTE) datasets systematically explore only a fraction of possible reagent, solvent, and catalyst combinations [41].
- Domain shifts: Models trained on general chemical databases often perform poorly when applied to specialized domains like organic photovoltaics or photocatalysis due to differing feature distributions [45] [42].
These limitations create a fundamental bottleneck in computational reaction mechanism research that requires specialized algorithmic approaches to overcome.
Transfer Learning Paradigms
Transfer learning approaches in chemistry can be categorized into several strategic frameworks:
- Model fine-tuning: Pre-trained models on large datasets (e.g., ChEMBL, USPTO) are adapted to specific reaction types with limited data. ReactionT5, a transformer model pre-trained on the Open Reaction Database, exemplifies this approach, achieving high performance in product prediction, retrosynthesis, and yield prediction even with limited fine-tuning data [46].
- Feature representation transfer: Learned chemical representations from source domains are transferred to target tasks. Graph convolutional networks (GCNs) pre-trained on virtual molecular databases have successfully predicted catalytic activity for real-world organic photosensitizers [45].
- Cross-domain knowledge transfer: Models transfer knowledge between different chemical domains. BERT models pre-trained on drug-like small molecules or chemical reactions have effectively predicted properties of organic materials, demonstrating transfer across seemingly disparate chemical domains [42].
The effectiveness of transfer learning hinges critically on the relationship between source and target domains. Studies on Pd-catalyzed cross-coupling reactions reveal that transfer learning works best when reaction mechanisms are closely related. For instance, models trained on benzamide nucleophiles successfully predicted outcomes for phenyl sulfonamide reactions (ROC-AUC = 0.928) but failed completely for pinacol boronate esters (ROC-AUC = 0.133), highlighting the mechanistic dependence of successful knowledge transfer [41].
Active Learning Strategies
Active learning frameworks address data scarcity through iterative, intelligent data acquisition:
- Uncertainty sampling: Selecting data points where model predictions are most uncertain [43].
- Diversity sampling: Choosing representatives from underrepresented regions of the chemical space.
- Expected model change: Prioritizing samples that would most alter the current model.
In reaction optimization, active learning iteratively selects the most promising reaction conditions to test experimentally. This approach has demonstrated superior outcomes compared to traditional human-driven experimentation, significantly streamlining the experimental process by identifying the most informative experiments from vast possibility spaces [43].
Methodological Approaches & Experimental Protocols
Integrated Active Transfer Learning Framework
The combination of transfer and active learning creates a powerful methodology for exploring new chemical reactions with limited data. The following workflow illustrates this integrated approach:
Figure 1: Active Transfer Learning Workflow for Reaction Optimization
This framework begins by leveraging existing knowledge from a source domain, then strategically acquires new experimental data in the target domain. The key advantage lies in using the transferred model to guide initial exploration, mimicking how expert chemists apply known reactions to new substrates [41].
Case Study: Pd-Catalyzed Cross-Coupling Optimization
A detailed experimental protocol demonstrates this framework applied to Pd-catalyzed cross-coupling reactions:
Phase 1: Model Pre-training and Transfer
- Source Data Curation: Collect high-throughput experimentation data for a related nucleophile type (e.g., 100+ data points for benzamide coupling) [41].
- Feature Engineering: Encode reaction components (electrophile, catalyst, base, solvent) using appropriate descriptors (Morgan fingerprints, molecular graphs, or physicochemical properties).
- Model Training: Train a random forest classifier or transformer model to predict binary reaction success (0% yield vs. >0% yield) using cross-validation to prevent overfitting.
- Model Transfer: Apply the trained model to the target nucleophile type, focusing on common reagent combinations between source and target domains.
Phase 2: Active Learning Cycle
- Initial Experimental Design: Use the transferred model to predict the 10-20 most promising reaction conditions for initial testing.
- High-Throughput Experimentation: Execute predicted reactions using nanomole-scale HTE in 1536-well plates to generate labeled data [41].
- Model Update: Fine-tune the transferred model with new experimental results.
- Informed Query Strategy: Apply uncertainty sampling to identify the most informative next experiments from the remaining condition space.
- Iterative Refinement: Repeat steps 2-4 for 3-5 cycles or until model performance plateaus.
This protocol achieved ROC-AUC scores up to 0.928 when transferring between mechanistically similar nucleophiles, demonstrating the power of the approach for related reaction types [41].
Case Study: Foundation Model Fine-tuning
An alternative protocol leveraging chemical foundation models:
Phase 1: Model Selection and Preparation
- Select Pre-trained Model: Choose a chemistry foundation model such as ReactionT5, pre-trained on large reaction databases (e.g., Open Reaction Database) [46].
- Task Formulation: Frame the target problem (yield prediction, condition recommendation) as a text-to-text task using reaction SMILES with role-specific tokens (e.g., "REACTANT:", "REAGENT:") [46].
Phase 2: Data-Efficient Fine-tuning
- Limited Data Sampling: Randomly select a small subset (50-200 reactions) from the target domain for fine-tuning.
- Progressive Training: Fine-tune the foundation model with a low learning rate (1e-5 to 1e-4) for limited epochs (10-50) to prevent catastrophic forgetting.
- Validation: Evaluate on held-out test sets from the target domain.
ReactionT5 achieved 97.5% accuracy in product prediction and 71.0% in retrosynthesis after limited fine-tuning, demonstrating strong performance even with small datasets [46].
Key Algorithms and Model Architectures
Model Selection Guidelines
Table 1: Machine Learning Models for Chemical Reaction Tasks with Limited Data
| Model Type | Best Suited Tasks | Data Requirements | Transfer Learning Capability | Performance Examples |
|---|---|---|---|---|
| Random Forest | Reaction condition classification, Yield prediction | 100+ data points | Feature representation transfer | ROC-AUC: 0.928 for C-N coupling [41] |
| Transformer (T5) | Product prediction, Retrosynthesis, Yield prediction | 50-200 fine-tuning examples | Full model fine-tuning | 97.5% product prediction accuracy [46] |
| Graph Neural Networks | Catalytic activity prediction, Molecular property prediction | 100+ data points | Pre-training on virtual libraries | Improved prediction of photosensitizer activity [45] |
| BERT-based Models | Virtual screening, Property prediction | 100+ fine-tuning examples | Unsupervised pre-training + fine-tuning | R² > 0.94 for HOMO-LUMO gap prediction [42] |
| QUBO Models | Reaction condition optimization, Large-scale screening | Varies with problem size | Adapted for active learning | Seconds for billions of condition screenings [43] |
Critical Implementation Considerations
Successful implementation requires attention to several algorithmic details:
- Model Simplification: For transfer learning between reaction types, simplified models with a small number of decision trees of limited depth enhance generalizability and interpretability while maintaining performance [41].
- Domain Similarity Assessment: Before transfer, evaluate mechanistic similarity between source and target reactions through:
- Reaction mechanism analysis (e.g., Pd-catalyzed C-N vs C-C coupling)
- Molecular similarity of key substrates (Tanimoto coefficients)
- Representation learning (embedding space distance)
- Active Learning Query Strategies: For reaction optimization, balance exploration (testing diverse conditions) and exploitation (refining promising conditions) using methods like ε-greedy or upper confidence bound [45].
Quantitative Performance Analysis
Benchmarking Results Across Reaction Types
Table 2: Transfer Learning Performance Across Different Chemical Domains
| Source Domain | Target Domain | Model Architecture | Performance Metric | Result | Key Insight |
|---|---|---|---|---|---|
| Benzamide Coupling | Phenyl Sulfonamide | Random Forest | ROC-AUC | 0.928 [41] | High transfer between mechanistically similar reactions |
| Benzamide Coupling | Pinacol Boronate | Random Forest | ROC-AUC | 0.133 [41] | Poor transfer across different reaction mechanisms |
| USPTO Reactions | Organic Photovoltaics | BERT | R² (HOMO-LUMO gap) | >0.94 [42] | Successful cross-domain transfer |
| Virtual Molecules | Organic Photosensitizers | Graph CNN | Predictive accuracy | Significantly improved [45] | Virtual libraries enhance real-world predictions |
| ORD Pre-training | Multiple Tasks | ReactionT5 | Product prediction accuracy | 97.5% [46] | Foundation models effective with limited fine-tuning |
| Baeyer-Villiger | Baeyer-Villiger (with transfer) | Transformer | Top-1 accuracy | 81.8% [44] | Marked improvement over baseline (58.4%) |
Impact of Data Volume on Model Performance
The relationship between dataset size and model performance follows characteristic patterns in limited-data regimes:
- Minimal viable data: For fine-tuning foundation models, 50-100 well-chosen examples often provide substantial improvements over the base model [46].
- Active learning returns: In reaction condition optimization, 3-5 cycles of active learning typically identify high-yielding conditions with 20-30% of the full experimental budget [43].
- Diminishing returns: Performance gains typically plateau after 200-300 targeted experiments for most reaction optimization tasks, guiding resource allocation decisions [41] [43].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Key Research Tools and Resources for Limited-Data Reaction Modeling
| Resource Name | Type | Primary Function | Application in Limited Data Scenarios |
|---|---|---|---|
| Open Reaction Database (ORD) | Data Resource | Large-scale reaction dataset | Pre-training foundation models like ReactionT5 [46] |
| USPTO Database | Data Resource | Patent-extracted reactions | Pre-training for cross-domain transfer learning [42] |
| ChEMBL | Data Resource | Bioactive molecule database | Pre-training for molecular property prediction [42] |
| RDKit | Software | Cheminformatics toolkit | Molecular descriptor calculation and fingerprinting [45] |
| ReactionT5 | Model | Chemical reaction foundation model | Fine-tuning for specific reactions with limited data [46] |
| Digital Annealer Unit | Hardware | Quantum-inspired optimizer | Rapid screening of billions of reaction conditions [43] |
| rxnfp | Software | BERT-based reaction framework | Reaction classification and yield prediction [42] |
Experimental Design Considerations
When applying these approaches to new reaction types, several practical considerations emerge:
- Data Quality over Quantity: In very low-data regimes (≤50 examples), careful curation of training examples significantly outperforms larger, noisier datasets [46].
- Transferability Assessment: Before extensive experimentation, evaluate potential source domains through:
- Mechanism similarity analysis
- Molecular structure alignment
- Preliminary cross-validation on available data
- Active Learning Initialization: When beginning active learning cycles, use domain-knowledge-informed initial points rather than purely random selection to accelerate convergence [41] [43].
The strategic integration of active and transfer learning methodologies provides a powerful framework for overcoming data scarcity in chemical reaction research. As demonstrated across multiple reaction types and optimization tasks, these approaches enable efficient navigation of complex chemical spaces while minimizing experimental resource requirements. The key principles for success include careful source domain selection based on mechanistic similarity, appropriate model architecture choices aligned with data constraints, and iterative experimental design that maximizes information gain per experiment.
Future directions in this field will likely involve more sophisticated foundation models pre-trained on expanded reaction databases, improved transferability metrics for predicting cross-domain performance, and tighter integration of computational prediction with automated experimental platforms. As these methodologies mature, they will increasingly accelerate the computational exploration of reaction mechanisms, particularly for novel and understudied reaction classes where traditional data-intensive approaches face fundamental limitations.
The computational exploration of chemical reaction mechanisms is fundamentally constrained by the exponential growth of possible reaction pathways, a phenomenon known as combinatorial explosion. This whitepaper examines cutting-edge computational strategies designed to tame this complexity, with a focus on the synergistic application of machine learning-driven energy filters and highly parallel computing architectures. Framed within the broader context of reaction mechanism research, we detail how these methodologies enable the efficient navigation of vast chemical reaction spaces, thereby accelerating discoveries in fields such as catalysis and pharmaceutical development. We present quantitative performance data, detailed experimental protocols, and essential toolkits to equip researchers with the practical knowledge to implement these advanced techniques.
In the computational exploration of chemical reaction networks, the number of possible intermediates and transition states grows exponentially with system size and reaction steps. This "exponential complexity" renders exhaustive quantum chemical screening intractable for all but the simplest transformations, presenting a significant bottleneck for the discovery and optimization of novel reactions, particularly in pharmaceutical and materials science.
Traditional approaches, which rely heavily on chemist intuition and one-factor-at-a-time (OFAT) optimization, are poorly suited to navigating these high-dimensional spaces. The field is therefore increasingly turning to a new paradigm that integrates data-driven machine learning (ML) filters to prune unlikely reaction paths and leverages parallel computing architectures—from classical high-throughput systems to emerging quantum frameworks—to evaluate promising candidates concurrently.
Core Methodologies and Quantitative Comparisons
Machine Learning as an Energy Filter
Machine learning models act as sophisticated energy filters by rapidly predicting the thermodynamic and kinetic feasibility of reaction steps, thereby identifying low-energy pathways for more computationally intensive verification.
Bayesian Optimization for Reaction Navigation: The Minerva framework exemplifies this approach, using Bayesian optimization to guide high-throughput experimentation (HTE). It balances the exploration of unknown reaction conditions with the exploitation of promising regions identified by a Gaussian Process (GP) regressor [47]. This model predicts reaction outcomes (e.g., yield, selectivity) and their uncertainties for vast sets of possible conditions, directing experimental resources toward optimal solutions.
Support Vector Regression for Sectoral Prediction: A genetic algorithm-optimized Support Vector Regression (SVR) model has been successfully applied to predict energy consumption and associated carbon emissions across residential, industrial, commercial, and agricultural sectors with high accuracy [48]. This demonstrates the utility of ML filters in managing complex, multi-parameter systems.
Table 1: Performance Metrics of Selected ML Filter Models
| Model | Application | Key Metric | Performance |
|---|---|---|---|
| GA-Optimized SVR [48] | Sectoral Energy Prediction | Mean Absolute Error (MAE) | Residential: 1.22%, Industrial: 4.98%, Commercial: 4.40%, Agricultural: 4.04% |
| R² | Approached 1 | ||
| Bayesian Optimization (Minerva) [47] | Ni-catalyzed Suzuki Reaction Optimization | Area Percent (AP) Yield / Selectivity | Identified conditions with 76% yield and 92% selectivity |
Parallel Computing Architectures
Parallel computing tackles exponential complexity by dividing the computational workload across many processing units, enabling the simultaneous evaluation of thousands of reaction conditions or pathway segments.
Highly Parallel High-Throughput Experimentation: The Minerva framework is integrated with automated HTE platforms that allow for the highly parallel execution of reactions—in batch sizes of 24, 48, or 96—within robotic platforms. This integration of ML with physical automation was key to navigating a space of 88,000 conditions for a nickel-catalyzed Suzuki reaction, a task where traditional chemist-designed plates failed [47].
Parallel Quantum Algorithms: For fundamental pathfinding problems, which are analogous to searching reaction networks, parallel Quantum Approximate Optimization Algorithm (QAOA) architectures have been proposed. This approach constructs two parallel QAOA circuits to simultaneously calculate connectivity energy and path energy for grid-based path planning, effectively mapping the problem to finding the minimum quantum energy state of an Ising model [49].
Table 2: Comparative Analysis of Parallel Computing Approaches
| Architecture | Application Context | Reported Acceleration/Performance |
|---|---|---|
| Classical HTE (Minerva) [47] | Chemical Reaction Optimization | Identified high-yield API synthesis conditions in 4 weeks vs. a previous 6-month campaign. |
| Parallel QAOA [49] | Grid Path Planning (Quantum) | Demonstrated capability to find optimal path coding combination with the highest probability using shallow (p=1) circuits. |
| Clustering-Based Pathfinding [50] | Shortest Path in Transport Graphs | 5x to 30x acceleration on real-world city graphs, with an average error of up to 15%. |
Detailed Experimental Protocols
Protocol: ML-Guided Bayesian Optimization Campaign for Reaction Optimization
This protocol is adapted from the Minerva framework for optimizing a chemical reaction with multiple objectives (e.g., yield and selectivity) [47].
Step 1: Define the Reaction Condition Space
- Action: Enumerate all plausible reaction parameters (e.g., ligands, solvents, catalysts, additives, concentrations, temperatures) based on chemical intuition and process constraints.
- Output: A discrete combinatorial set of potential reaction conditions, automatically filtered to exclude impractical or unsafe combinations (e.g., temperatures exceeding solvent boiling points).
Step 2: Initial Quasi-Random Sampling
- Action: Use algorithmic Sobol sampling to select an initial batch of experiments (e.g., a 96-well plate).
- Rationale: This maximizes the initial coverage of the reaction space, increasing the probability of discovering informative regions.
Step 3: Automated High-Throughput Execution
- Action: Execute the batch of reactions using an automated HTE robotic platform.
- Analysis: Analyze reaction outcomes (e.g., via UPLC/GC) to obtain quantitative data for objectives like yield and selectivity.
Step 4: Train Machine Learning Model and Select Next Batch
- Action: Train a Gaussian Process (GP) regressor on all accumulated experimental data to predict reaction outcomes and their uncertainties for all possible conditions in the defined space.
- Action: Use a scalable multi-objective acquisition function (e.g., q-NParEgo, TS-HVI) to evaluate all conditions and select the next most promising batch of experiments. This function balances exploring uncertain regions of the space and exploiting known high-performing regions.
Step 5: Iterate and Converge
- Action: Repeat Steps 3 and 4 for multiple iterations.
- Termination: The campaign is terminated upon convergence (no significant improvement), identification of a satisfactory solution, or exhaustion of the experimental budget.
Protocol: Model Complexity Reduction for Agent-Based Reaction Simulations
This protocol outlines methods for reducing the computational complexity of fine-grained agent-based models (ABMs) of chemical reactions, as demonstrated in a study of a simple A + B → C system [51].
Step 1: Establish the Baseline Model
- Action: Implement a full-scale ABM where each molecule is represented as a distinct agent moving via Brownian motion in a bounded volume. Interactions occur when agents are within a specified interaction range.
Step 2: Apply Complexity Reduction Techniques
- Scale Reduction:
- Action: Reduce the total number of agents by a scale factor (e.g., 10x).
- Compensation: Increase the interaction range of each agent by the cube root of the scale factor (
range = baseRange × (1/modelScale)^(1/3)) to maintain a similar probability of interaction per unit time.
- Time Step Increase:
- Action: Increase the length of each simulation iteration.
- Compensation: Increase the agent interaction range by the cube root of the time step increase (
range = baseRange × (timeStep)^(1/3)) and adjust the random walk step distance proportionally to the square root of the time step (distance = √(diffusion × timestep)).
- Scale Reduction:
Step 3: Validate and Benchmark
- Action: Run multiple simulations of the reduced-complexity models and the baseline model from randomized starting conditions.
- Analysis: Compare the system-level outputs (e.g., the rate of depletion of reactants A and B) and runtime performance to quantify trade-offs between accuracy, variance, and computational efficiency.
Visualization of Workflows and Logical Relationships
Diagram: ML-Driven Reaction Optimization Workflow
The following diagram illustrates the iterative Design-Build-Test-Learn (DBTL) cycle implemented in the Minerva framework [47].
Diagram: Parallel QAOA for Pathfinding
This diagram outlines the parallel QAOA circuit architecture proposed for quantum path planning, which is analogous to searching for optimal pathways in a reaction network [49].
The Scientist's Toolkit: Essential Research Reagents & Solutions
This table details key components used in the advanced computational and experimental methodologies discussed herein.
Table 3: Key Research Reagents and Computational Tools
| Item / Solution | Function / Role | Application Context |
|---|---|---|
| Minerva Framework | A scalable machine learning framework for highly parallel, multi-objective reaction optimisation. | Integrates Bayesian optimisation with HTE to navigate large chemical spaces [47]. |
| Gaussian Process (GP) Regressor | A probabilistic ML model that predicts reaction outcomes and quantifies prediction uncertainty. | Serves as the surrogate model in Bayesian optimisation to guide experimental design [47]. |
| Bayesian Optimization | An acquisition function that balances exploration and exploitation to select the next experiments. | Core to ML-driven workflows like Minerva for efficiently finding optimal conditions [47]. |
| RxnNet Platform | An AI-assisted platform that integrates heuristic chemical rules with quantum chemistry to automate the discovery of reaction mechanisms. | Used for constructing mechanistically informed reaction networks, e.g., in carbocation chemistry [5]. |
| Quantum Approximate Optimization Algorithm (QAOA) | A hybrid quantum-classical algorithm for solving combinatorial optimization problems. | Applied to pathfinding problems by mapping them to the problem of finding a quantum ground state [49]. |
| Contraction Hierarchies (CH) | A graph preprocessing technique that iteratively contracts less important nodes to create shortcuts. | Accelerates shortest-path queries in graph-based problems, such as transport networks [50]. |
| FLAME Framework | A platform for developing and executing high-performance, parallel agent-based models. | Used for simulating complex systems, including molecular-level chemical reactions [51]. |
| Non-Precious Metal Catalysts (e.g., Nickel) | Earth-abundant catalysts that reduce cost and environmental impact compared to precious metals. | Target for optimisation in campaigns like the Ni-catalyzed Suzuki reaction [47]. |
The application of machine learning (ML) in chemistry has revolutionized the prediction of reaction outcomes and the exploration of chemical space. However, a significant performance gap persists for reactions involving transition metals and catalytic cycles, which are pillars of modern synthetic chemistry. These systems present unique challenges, including complex electron interactions, multi-step mechanisms, and the scarcity of standardized, high-quality data, which hinder the generalizability of ML models trained primarily on organic reactants [52]. This domain gap limits our ability to fully leverage artificial intelligence for the computational exploration of chemical reaction mechanisms, particularly in areas like drug development where metal-catalyzed reactions are increasingly relevant.
Bridging this gap requires a multi-faceted approach, combining innovative model architectures that incorporate physical constraints, advanced data generation strategies, and specialized validation protocols. This guide synthesizes current research to provide a technical roadmap for developing more robust and reliable models capable of handling the complexity of metallic systems and catalytic processes, thereby accelerating discovery in pharmaceutical and materials science.
The Unique Complexities of Metal-Catalyzed Reactions
Transition metal-catalyzed reactions are intrinsically more complex than their purely organic counterparts, which creates fundamental challenges for ML models. The core issue lies in the high dimensionality of the tunable parameters and the intricate interplay of steric, electronic, and mechanistic factors that govern catalytic activity and selectivity [52].
Unlike organic molecules where bonding is often straightforward, metal complexes exhibit characteristics that are difficult to represent with standard molecular descriptors. These include variable coordination geometries, redox-active metal centers, and complex ligand-field effects. Furthermore, catalytic cycles involve multiple interconnected intermediates and transition states, making the reaction landscape vast and rugged. The scarcity of standardized, high-quality experimental data for these systems exacerbates the problem, as ML models are inherently data-hungry. This data scarcity is not merely a question of volume but also of diversity; many existing datasets underrepresent the broad spectrum of metals, ligands, and reaction types used in practical catalysis [52] [8].
Foundational Strategies for Model Improvement
Incorporating Physical Principles into Model Architecture
A primary reason for model failure in unfamiliar chemical domains is the lack of embedded physical constraints. Models that treat chemistry as a purely statistical problem can generate predictions that are mathematically plausible but physically impossible, such as violating the conservation of mass.
- Electron-Conservative Models: Cutting-edge approaches are now addressing this by grounding predictions in fundamental physical principles. For instance, the FlowER (Flow matching for Electron Redistribution) model developed at MIT uses a bond-electron matrix, a method rooted in 1970s chemistry, to explicitly represent the electrons in a reaction. This representation ensures the model conserves both atoms and electrons throughout the predicted reaction process, preventing the generation of "alchemical" outcomes where atoms spuriously appear or disappear [8].
- Neural Network Potentials (NNPs): For accurately modeling the potential energy surfaces of catalytic systems, NNPs like the EMFF-2025 framework offer a powerful tool. These models are trained on quantum mechanical data (e.g., from Density Functional Theory calculations) and can achieve near-DFT accuracy in predicting structures, mechanical properties, and reaction characteristics at a fraction of the computational cost. This is particularly valuable for simulating the decomposition mechanisms and stability of complex molecular systems under various conditions [53].
Advanced Data Generation and Curation Techniques
Overcoming data scarcity requires proactive and intelligent data generation. Relying solely on existing literature data, which is often biased toward successful reactions, is insufficient for training robust models.
- Transfer Learning: This strategy has proven highly effective for optimizing ML models with minimal new data. The development of the general-purpose EMFF-2025 NNP for high-energy materials (HEMs) demonstrates this: researchers started with a pre-trained model (DP-CHNO-2024) and refined it for new material systems by incorporating a small amount of targeted DFT calculation data. This approach leverages existing knowledge, reducing the need for extensive new training and improving performance on related but distinct chemical systems [53].
- Chemical Reaction Network (CRN) Analysis: To gain insight into complex reaction pathways, such as those in sol-gel synthesis for metal-organic frameworks, a data-driven CRN approach can be employed. This involves using text-mined data from scientific literature to identify successful synthesis trends and then building a mathematical network of possible chemical species and reactions. Computational tools like HiPRGen (High Performance Reaction Generation) and Reaction Network Monte Carlo (RNMC) are then used to explore this network, predicting feasible reaction pathways and intermediates that can be validated and added to training datasets [54].
- Text-Mining for Synthesis Protocols: Systematically analyzing hundreds of scientific publications allows for the extraction of synthesis parameters and outcomes. For example, a study on BiFeO3 formation analyzed 340 synthesis recipes to identify that nitrate salts and 2-methoxyethanol as a solvent frequently lead to the desired phase-pure outcome. This curated, high-quality dataset provides a solid foundation for modeling complex inorganic syntheses [54].
Table 1: Quantitative Performance of Advanced Modeling Strategies
| Strategy | Model/Technique | Reported Performance | Key Application Domain |
|---|---|---|---|
| Physical Principle Integration | FlowER (MIT) | Massive increase in prediction validity & conservation; matching or better accuracy vs. benchmarks [8] | General reaction prediction |
| Neural Network Potentials | EMFF-2025 NNP | Mean Absolute Error (MAE): Energy ±0.1 eV/atom, Force ±2 eV/Å [53] | High-energy materials (C, H, N, O) |
| Data Generation & Curation | Transfer Learning (DP-GEN framework) | Accurately predicts surface reconstruction & segregation without explicit surface data in training [53] | Material surfaces and nanoalloys |
Visualization of an Integrated Workflow for Model Improvement
The following diagram illustrates a synergistic workflow that combines the strategies outlined above to systematically address domain gaps in modeling metal catalysis.
Experimental Protocols for Validation and Benchmarking
Rigorous validation is critical to ensure that improvements in model performance are genuine and not artifacts of a limited test set. The following protocols provide a framework for benchmarking models on metal and catalytic cycle tasks.
Protocol 1: Mechanistic Pathway Validation
Objective: To evaluate a model's ability to predict not just the final products, but also the correct intermediate steps of a catalytic cycle.
- Dataset Curation: Assemble a benchmark dataset of catalytic reactions with well-established mechanisms, such as palladium-catalyzed cross-couplings or ruthenium-catalyzed metatheses. The dataset should include explicit representations of key intermediates (e.g., oxidative addition, transmetalation, reductive elimination products) [52] [8].
- Model Prediction: Use the candidate model to predict the reaction pathway from the initial reactants and catalyst.
- Analysis: Compare the predicted intermediates and transition states against the known mechanistic pathway. Key metrics include:
- Pathway Accuracy: The percentage of correctly identified intermediate species.
- Electron Conservation: The rate at which the model's proposed steps conserve electrons and atom count [8].
- Energy Profile Plausibility: For models with energy output, assess whether the relative energies of intermediates align with established thermodynamic and kinetic principles.
Protocol 2: Generalization to Unseen Metal Centers
Objective: To test a model's capacity to make accurate predictions for metals that were not present in its training data.
- Data Splitting: Partition a dataset of catalytic reactions by the identity of the transition metal center (e.g., Pd, Ni, Ru, Cu, Fe). Ensure that one or more entire metal categories are held out from the training set.
- Model Training & Evaluation: Train the model on the training set and evaluate its performance on the hold-out set containing unseen metals.
- Analysis: Measure standard metrics (e.g., MAE, R²) for predicting reaction yields or selectivity on the unseen metals. A significant performance drop indicates poor generalization, highlighting the need for better descriptor sets or model architectures that can capture periodic trends. The use of principal component analysis (PCA) and correlation heatmaps can help visualize whether the model is effectively organizing the chemical space of different metals [53].
Table 2: Key Reagents and Computational Tools for Research
| Reagent / Tool Name | Type | Function in Research |
|---|---|---|
| Nitrate Salts | Chemical Precursor | Common, effective metal source in sol-gel synthesis; promotes desired oligomerization pathways for pure phase formation [54]. |
| 2-Methoxyethanol (2ME) | Solvent | Dominant solvent in many metal-organic syntheses; stabilizes de-nitrated metal complexes and facilitates key dimerization reactions [54]. |
| Citric Acid | Chelating Agent | Additive that frequently leads to phase-pure product formation in complex metal oxides by modulating metal ion reactivity [54]. |
| HiPRGen & RNMC | Software Tools | Generate and traverse Chemical Reaction Networks (CRNs) to explore possible reaction pathways and intermediates computationally [54]. |
| DP-GEN Framework | Software Tool | Automated workflow for generating general-purpose Neural Network Potentials (NNPs) via active learning and transfer learning [53]. |
| Bond-Electron Matrix | Representational Framework | Ensures physical constraints (mass/electron conservation) are baked into reaction prediction models, as used in FlowER [8]. |
Addressing the domain gaps in machine learning models for metal-containing systems and catalytic cycles is a central challenge in the computational exploration of chemical reaction mechanisms. Success hinges on moving beyond purely data-driven approaches and instead building models that are informed by fundamental physical chemistry, augmented by strategic data generation, and rigorously validated against chemically meaningful benchmarks. The integration of electron-conservative architectures, transfer learning with NNPs, and data mining from chemical reaction networks represents a powerful, synergistic strategy.
The future of this field lies in the continued development of self-driving laboratories that combine human expertise, AI, and robotics. In this paradigm, the models described here will not only predict reactions but also design and execute experiments to validate their own predictions, particularly targeting the most uncertain areas of chemical space related to catalysis. For drug development professionals, the ultimate goal is to have reliable, predictive tools that can accurately model the complex metal-catalyzed reactions used in late-stage functionalization and the synthesis of chiral active pharmaceutical ingredients, thereby streamlining the entire drug discovery pipeline.
In the computational exploration of chemical reaction mechanisms, researchers face a fundamental compromise: the need for high-throughput screening of potential reaction pathways against the demand for high-accuracy energetics and kinetics. Semiempirical quantum chemical methods like GFN2-xTB offer remarkable speed, enabling the investigation of large molecular systems and extensive reaction networks that would be prohibitively expensive with density functional theory (DFT). However, this computational efficiency comes with well-documented limitations in accuracy, particularly for reaction barriers, transition metal complexes, and systems prone to delocalization error [55] [56]. This technical guide examines strategies for strategically integrating GFN2-xTB and DFT to create efficient yet reliable workflows for reaction mechanism research and drug development.
The core challenge lies in method selection based on specific research objectives. GFN2-xTB operates within a parameterized tight-binding framework, producing molecular geometries and noncovalent interactions rapidly enough for large-scale conformer searches, implicit solvent calculations, or molecular dynamics runs where DFT would be prohibitive [56]. Nevertheless, GFN2-xTB and other low-cost methods have recognized limitations: reaction barriers often appear too low, orbital gaps become compressed, and transition-metal complexes can sometimes distort into unphysical geometries [56]. Understanding these systematic biases enables researchers to deploy GFN2-xTB where its approximations remain valid while reserving computationally intensive DFT calculations for critical validation steps.
Quantitative Method Comparison: Performance Benchmarks
A rigorous evaluation of method performance across chemically relevant benchmarks provides the foundation for developing balanced workflows. The following tables summarize key performance metrics for GFN2-xTB, next-generation methods, and DFT for common computational tasks in reaction mechanism research.
Table 1: Overall Performance Benchmark on GMTKN55 Database (≈32,000 Relative Energies)
| Method | WTMAD-2 (kcal/mol) | Computational Cost | Key Strengths |
|---|---|---|---|
| GFN2-xTB | 25.0 | Very Low | High-speed screening, large systems |
| g-xTB | 9.3 | Low | Improved thermochemistry/barriers |
| NN-xTB | 5.58 | Low | DFT-like accuracy, near-xTB speed |
| DFT (Typical) | ~5-15 | High to Very High | Gold-standard accuracy |
Table 2: Performance for Specific Chemical Applications
| Application | GFN2-xTB Performance | Recommended Method | Notes |
|---|---|---|---|
| Proton Transfer Barriers [57] | MUE: 13.5 kJ/mol | PM7 (13.4) or DFT | Varies by chemical group |
| Thia-Michael Reaction [55] | Incorrect potential surface | ωB97X-3c or good NNPs | Fails due to delocalization error |
| Bond Dissociation Energies [56] | MAE: 7.88 kcal/mol | g-xTB (MAE: 3.96 kcal/mol) | After linear correction |
| Protein-Ligand Interactions [56] | Moderate accuracy | g-xTB | Outperforms some NNPs |
| Infrared Spectroscopy [58] | <10% error (central frequencies) | GFN2-xTB sufficient | Reasonable for liquid solutions |
The benchmark data reveals a clear performance hierarchy. For general thermochemical accuracy across diverse chemical space (GMTKN55), GFN2-xTB shows substantial errors (25.0 kcal/mol WTMAD-2) compared to its successor g-xTB (9.3 kcal/mol) and neural-network enhanced NN-xTB (5.58 kcal/mol) [56] [59]. For specific reaction types like the thia-Michael reaction—relevant to covalent inhibitor design—GFN2-xTB fails to correctly describe the potential energy surface due to inadequate treatment of electron delocalization, a limitation addressed by range-separated hybrid functionals or carefully validated neural network potentials [55].
Integrated Workflow Design: Strategic Method Integration
The quantitative benchmarks support a stratified approach where methods are selected based on the specific task within the reaction mechanism exploration pipeline. The following workflow diagram illustrates this integrated strategy:
Diagram 1: Reaction exploration workflow.
This workflow leverages the complementary strengths of different computational methods. GFN2-xTB or the more accurate g-xTB serves as a filtering tool to identify plausible reaction pathways from a vast possibility space. Promising candidates then advance to DFT validation, initially through single-point energy calculations on the semiempirical geometries—a cost-effective strategy that captures the primary electronic energy contributions without the expense of full DFT geometry optimization. Only the most mechanistically significant pathways undergo comprehensive DFT analysis.
For complex reaction networks involving multiple intermediates and transition states, automated exploration tools like ARplorer demonstrate how this integration can be systematized. ARplorer combines GFN2-xTB for rapid potential energy surface generation with Gaussian's algorithms for transition state searching, creating a hybrid workflow that maintains efficiency while improving accuracy [4]. The program employs active-learning methods in transition state sampling and parallel multi-step reaction searches with efficient filtering to enhance efficiency and accelerate potential energy surface searching [4].
Advanced Protocols: Method-Specific Experimental Procedures
Sparse Identification from Limited Concentration Profiles
For experimental data analysis, a sparse identification approach can determine reaction mechanisms from limited concentration profiles, providing accurate kinetic models while preventing overfitting [60] [61]. This methodology is particularly valuable for reactions involving untraceable intermediates.
Procedure:
- Data Collection: Monitor temporal concentration profiles of traceable species (e.g., via UV-vis absorption spectroscopy) under multiple initial conditions [61].
- Intermediate Assumption: Postulate potential intermediate compositions based on known compounds and chemical reasoning.
- Model Generation: Enumerate all possible elementary steps (1-2 reactants) satisfying mass conservation.
- Sparse Regression: Apply L1-regularized regression (LASSO) to rate constant optimization in logarithmic space (x = log₁₀(k + 1)) to eliminate steps with negligible rate constants.
- Model Validation: Compare simulated concentration profiles against experimental data using mean relative squared error (MRSE), with cross-validation between training and test sets [61].
Application: This approach successfully identified 11 elementary steps involving 8 chemical species for the autocatalytic reduction of manganese oxide ions, using only concentration profiles of two manganese species [61].
LLM-Guided Automated Reaction Pathway Exploration
The ARplorer program implements a sophisticated protocol for automated reaction pathway discovery that integrates quantum mechanics and rule-based methodologies, underpinned by large language model-assisted chemical logic [4].
Procedure:
- Active Site Identification: Identify active sites and potential bond-breaking locations to setup multiple input molecular structures.
- Structure Optimization: Optimize molecular structures through iterative transition state searches using active-learning sampling.
- IRC Analysis: Perform intrinsic reaction coordinate analysis to derive new reaction pathways.
- Pathway Completion: Eliminate duplicate pathways and finalize structures for subsequent iterations.
- Chemical Logic Application: Apply both general chemical logic from literature and system-specific chemical logic from specialized LLMs to filter unlikely pathways [4].
Method Integration: The protocol uses GFN2-xTB for rapid potential energy surface generation with the option to employ DFT for more precise calculations when necessary [4].
Research Reagent Solutions: Computational Tools for Reaction Exploration
Table 3: Essential Computational Tools for Reaction Mechanism Research
| Tool/Resource | Function | Application Context |
|---|---|---|
| GFN2-xTB | Semiempirical electronic structure | Initial pathway screening, large systems |
| g-xTB | Next-generation tight-binding | Improved accuracy with minimal speed cost |
| NN-xTB | Machine-learning enhanced xTB | Near-DFT accuracy, maintains electronic structure |
| ωB97X-3c | Range-separated hybrid DFT | Problematic systems (e.g., thia-Michael) |
| ARplorer | Automated pathway exploration | Systematic reaction network mapping |
| CETSA | Experimental target engagement | Validation in biological systems [38] |
The computational toolkit spans multiple levels of theory, each with distinct advantages. GFN2-xTB remains valuable for initial screening where its speed enables investigation of large chemical spaces. The newer g-xTB method addresses several GFN2-xTB limitations through a charge-dependent, polarization-capable basis set and range-separated approximate Fock exchange, dramatically improving performance for reaction barriers and orbital gaps [56]. For the highest accuracy requirements, range-separated hybrid functionals like ωB97X-3c correctly describe challenging electronic environments where GFN2-xTB fails, such as the thia-Michael reaction relevant to covalent inhibitor design [55].
Application Case Studies: From Theory to Practice
Covalent Inhibitor Warhead Reactivity
The thia-Michael reaction between thiolates and α,β-unsaturated carbonyls provides a critical test case for computational method selection. These reactions are fundamental to covalent inhibitor design, with electrophilic warheads targeting cysteine residues in therapeutic compounds like ibrutinib [55].
Challenge: Standard density functionals without sufficient exact exchange (PBE, B3LYP) fail to correctly model this reaction, favoring long-range anion-to-π* charge transfer instead of the carbanion product [55].
Solution Strategy:
- Initial Screening: Use GFN2-xTB for rapid assessment of multiple warhead scaffolds.
- Validation: Apply range-separated hybrid functionals (ωB97X-3c) for accurate energetics.
- Alternative: Employ validated neural network potentials (eSEN-OMol25-sm-conserving, UMA-S) for near-DFT accuracy at significantly reduced computational cost [55].
This case illustrates the danger of relying exclusively on GFN2-xTB for systems with specific electronic structure requirements, while demonstrating how hybrid workflows maintain efficiency without sacrificing accuracy.
Complex Reaction Network Elucidation
For multi-step reactions with parallel pathways and elusive intermediates, a layered computational approach becomes essential. The autocatalytic reduction of permanganate by oxalic acid exemplifies this challenge, with multiple manganese oxidation states and transient intermediates [61].
Experimental Constraint: Only Mn⁷⁺ and Mn³⁺ concentrations were directly measurable via UV-vis spectroscopy.
Computational Strategy:
- Sparse Identification: Applied L₁-regularized regression to select essential elementary steps from 64 possible candidates.
- Intermediate Hypothesis: Assumed five possible intermediates (Mn⁶⁺, Mn⁵⁺, Mn⁴⁺, C₂O₄⁻, CO₂⁻) based on chemical reasoning.
- Mechanism Reduction: Identified 11 core elementary steps involving 8 chemical species that sufficiently reproduced experimental data [61].
This approach demonstrates how computational methods can extract detailed mechanistic information from limited experimental data through appropriate constraint of the solution space.
The computational exploration of chemical reaction mechanisms requires thoughtful method selection rather than universal reliance on either high-speed approximate methods or high-accuracy intensive calculations. GFN2-xTB remains a valuable tool for initial screening and large-system exploration, particularly when used with awareness of its systematic limitations. The emergence of improved successors like g-xTB and enhanced versions like NN-xTB narrows the accuracy gap while maintaining computational efficiency.
For research contexts requiring high confidence in results—such as drug development projects or detailed mechanistic proposals—strategic integration with DFT validation remains essential. The workflows and protocols outlined provide a framework for achieving this balance, enabling researchers to accelerate discovery while maintaining scientific rigor in computational reaction mechanism studies.
Benchmarking Digital Discovery: Validating and Comparing Computational Tools
The computational exploration of chemical reaction mechanisms is a cornerstone of modern chemical research, driving advances in drug discovery, catalyst design, and materials science. As the field increasingly adopts machine learning (ML) and other advanced computational techniques, the rigorous evaluation of these methods has become paramount. Assessing predictive accuracy, computational cost, and validity rates forms the critical triad that determines the real-world utility and reliability of computational models in chemical sciences. Without standardized evaluation frameworks, comparing methods and translating computational findings into laboratory successes remains challenging. This guide provides an in-depth technical examination of evaluation metrics and methodologies, offering researchers a structured approach to validate their computational tools within the context of chemical reaction mechanism research. By establishing consistent evaluation criteria, the scientific community can accelerate the development of more robust, efficient, and trustworthy computational models that effectively bridge the gap between theoretical prediction and experimental validation.
Evaluating Predictive Accuracy in Chemical Models
Predictive accuracy stands as the primary metric for assessing how well computational models replicate or forecast chemical phenomena. In chemical reaction mechanism research, this evaluation extends beyond simple error measurements to encompass specialized metrics that account for uncertainty, data imbalance, and domain-specific requirements.
Foundational Accuracy Metrics
The assessment of predictive accuracy begins with foundational statistical metrics that quantify the discrepancy between predicted and observed values. For regression tasks common in predicting reaction energies, barrier heights, or spectroscopic properties, the Mean Absolute Error (MAE) and Root Mean Squared Error (RMSE) provide straightforward measures of deviation. When working with limited data, expressing RMSE as a percentage of the target value range (scaled RMSE) facilitates interpretation across different chemical systems [62]. For classification problems such as predicting reaction occurrence or regioselectivity, standard metrics include accuracy, precision, recall, and F1-score. However, these conventional metrics often prove inadequate for the imbalanced datasets typical in chemical research, where inactive compounds vastly outnumber active ones or certain reaction outcomes are rare [63].
Uncertainty-Aware Evaluation
For probabilistic models that provide uncertainty estimates alongside predictions, specialized metrics are essential. The area metric (AM) quantifies the disagreement between the cumulative distribution functions of predictions and observations, capturing both accuracy (bias) and precision (variance) components [64]. This metric can be decomposed into absolute bias (related to accuracy) and uncertainty width (related to precision), enabling researchers to diagnose whether poor performance stems from systematic error or excessive uncertainty [64]. Gaussian Process (GP) models naturally provide uncertainty quantification, with performance evaluated using metrics like the standard R² coefficient of determination and examination of prediction confidence intervals [65].
Domain-Specific Accuracy Measures
Chemical applications often require specialized accuracy metrics aligned with research goals. Precision-at-K measures the model's ability to identify the most promising candidates (e.g., top catalysts or drug candidates) from a ranked list, crucial for virtual screening workflows [63]. Rare event sensitivity evaluates performance on low-frequency but critical occurrences, such as predicting toxicological signals or rare reaction pathways [63]. Pathway impact metrics assess whether predictions align with biologically relevant pathways or reaction mechanisms, ensuring biological or chemical interpretability beyond statistical correctness [63].
Table 1: Metrics for Evaluating Predictive Accuracy in Chemical Models
| Metric Category | Specific Metrics | Application Context | Interpretation Guidelines |
|---|---|---|---|
| Foundational Regression | MAE, RMSE, Scaled RMSE, R² | Predicting continuous properties (energy, yield, etc.) | Lower values indicate better performance; R² closer to 1.0 preferred |
| Foundational Classification | Accuracy, Precision, Recall, F1-score | Binary outcomes (reaction success/failure, selectivity) | Consider dataset balance; F1-score balances precision and recall |
| Uncertainty Quantification | Area Metric (AM), Uncertainty Width | Probabilistic models (Gaussian Processes, Bayesian NN) | AM captures total disagreement; decomposition diagnoses error sources |
| Domain-Specific | Precision-at-K, Rare Event Sensitivity, Pathway Impact | Virtual screening, toxicity prediction, mechanistic modeling | Focuses on practically relevant predictions rather than overall performance |
Assessing Computational Cost and Efficiency
Computational cost directly impacts the feasibility and scalability of research in chemical reaction mechanisms. A comprehensive assessment requires evaluating multiple dimensions of resource utilization across different modeling approaches and experimental scales.
Metrics for Computational Efficiency
The most direct computational cost metrics include training time (for ML models), inference time (time required for prediction), and speed-up factors relative to reference methods [65]. For example, Gaussian Process models applied to H₂/air auto-ignition case studies demonstrated speed-up factors of 1.9-2.1 relative to direct integration of differential equations, while Artificial Neural Networks achieved factors up to 3.0 [65]. Memory and storage requirements present additional constraints, particularly for large-scale quantum chemistry calculations or deep learning models with millions of parameters. Scalability analysis examines how these resource demands increase with system size (atoms, electrons) or data volume, often characterized using Big O notation.
Data Efficiency Considerations
Data efficiency—the amount of training data required to achieve target performance levels—represents a critical aspect of computational cost, especially in low-data regimes common in experimental chemistry. Studies demonstrate that probabilistic methods like Gaussian Process Regression (GPR) and Gaussian Process Autoregressive Regression (GPAR) can outperform Artificial Neural Networks (ANNs) when working with small datasets, achieving R² values of 0.997-0.998 compared to 0.988 for ANNs on chemical source term prediction tasks [65]. The learning curve (performance as a function of training set size) provides a comprehensive view of data efficiency, revealing diminishing returns points that inform optimal data collection strategies.
Workflow Efficiency and Automation
Recent advances focus on automating computational workflows to enhance efficiency. Tools like ARplorer integrate quantum mechanics with rule-based approaches and large language model-guided chemical logic to automate reaction pathway exploration, significantly reducing human intervention [4]. The ROBERT software provides automated workflows for machine learning in low-data regimes, performing data curation, hyperparameter optimization, and model selection with minimal user input [62]. These automated systems employ active learning strategies that iteratively select the most informative data points for calculation, maximizing information gain while minimizing computational expense.
Table 2: Computational Cost Metrics for Chemical Modeling
| Cost Dimension | Evaluation Metrics | Measurement Approaches | Representative Values |
|---|---|---|---|
| Time Efficiency | Training time, Inference time, Speed-up factor | Comparison to reference methods (e.g., direct integration) | GP models: 1.9-2.1x speed-up; ANN: up to 3.0x speed-up [65] |
| Resource Requirements | Memory usage, Storage needs, CPU/GPU hours | Profiling tools, system monitoring | Dependent on system size and method complexity |
| Data Efficiency | Learning curves, Performance vs. dataset size | Progressive sampling and evaluation | GPR/GPAR outperform ANNs on small datasets (R²: 0.997-0.998 vs 0.988) [65] |
| Automation Impact | Human time reduction, Error rate reduction, Iteration cycle time | Comparison to manual approaches | ARplorer automates multi-step reaction pathway exploration [4] |
Determining Validity and Reliability
Validity assessment ensures that computational predictions are not only statistically sound but also chemically meaningful and reliable for guiding experimental work. This requires evaluating both technical correctness and domain relevance.
Statistical Validation Methods
Robust statistical validation forms the foundation of validity assessment. Cross-validation techniques, particularly in low-data regimes, must account for both interpolation and extrapolation performance. The selective sorted k-fold approach, which partitions data based on target value sorting and considers the highest RMSE between top and bottom partitions, provides rigorous extrapolation assessment [62]. Y-shuffling (randomizing target values) and one-hot encoding tests help identify potentially flawed models that capture spurious correlations [62]. For Bayesian models, examination of calibration curves determines whether reported confidence intervals accurately reflect empirical error rates.
Domain-Relevant Validation
Beyond statistical measures, domain-specific validation ensures chemical plausibility. Mechanistic consistency checks whether predictions align with established chemical principles and reaction mechanisms. Template-based validation, exemplified by tools like ARplorer, uses chemical logic encoded as SMARTS patterns to filter unlikely reaction pathways [4]. Experimental concordance measures agreement with laboratory observations, serving as the ultimate validity test. The integration of multi-fidelity validation—comparing predictions across different theoretical levels (e.g., semiempirical, DFT, and coupled-cluster methods)—provides insights into method dependency and systematic errors.
Addressing Overfitting in Limited Data Environments
Overfitting presents a particular challenge in chemical research where data is often scarce. Combating this requires specialized techniques during model development and validation. The ROBERT workflow employs a combined RMSE metric during hyperparameter optimization that incorporates both interpolation (10× 5-fold cross-validation) and extrapolation (selective sorted 5-fold CV) performance [62]. Regularization strategies—including L1/L2 regularization for linear models, dropout for neural networks, and complexity penalties for tree-based methods—constrain model flexibility to improve generalization. Benchmarking against simple baselines (e.g., y-mean prediction) provides a reality check for apparent model performance [62].
Integrated Workflows for Comprehensive Model Assessment
Comprehensive model evaluation requires integrated workflows that simultaneously address predictive accuracy, computational cost, and validity considerations. These workflows provide structured approaches for method selection and performance assessment across diverse chemical applications.
Automated Evaluation Workflows
Automated evaluation systems streamline the assessment process while reducing human bias. The ROBERT scoring system implements a comprehensive 10-point scale that weights predictive ability and overfitting (8 points), prediction uncertainty (1 point), and detection of spurious predictions (1 point) [62]. This system evaluates multiple performance aspects including cross-validation and test set performance using scaled RMSE, overfitting detection through performance differences, extrapolation ability, prediction consistency, and robustness to data permutations. Such automated assessment enables objective comparison across different algorithmic approaches and informs model selection for specific chemical applications.
Case Study: Reaction Pathway Exploration
The ARplorer program exemplifies an integrated approach to reaction mechanism evaluation, combining quantum mechanical methods with rule-based filtering and LLM-guided chemical logic [4]. Its workflow includes active site identification, transition state sampling through active learning, intrinsic reaction coordinate analysis, and pathway filtering using both general and system-specific chemical logic. This integrated design addresses all three evaluation dimensions: predictive accuracy through QM calculations, computational cost via efficient sampling and parallelization, and validity through chemical logic implementation. The system demonstrates how automated tools can balance rigorous evaluation with practical efficiency in complex reaction space exploration.
Specialized Evaluation for Chemical Applications
Domain-specific evaluation frameworks address the unique requirements of chemical applications. For regio- and site-selectivity prediction, evaluation must account for the distinction between these related concepts—site-selectivity refers to reactions at defined positions among identical options, while regioselectivity concerns orientation preferences during bond formation [66]. Specialized benchmarks and datasets have emerged for these applications, enabling standardized comparison across methods. In drug discovery, evaluation emphasizes early recognition performance through metrics like enrichment factors and precision-recall curves that reflect the practical goal of identifying active compounds amid large chemical libraries [63].
Model Evaluation Workflow: This diagram illustrates the integrated process for comprehensive model assessment, incorporating accuracy, cost, and validity evaluation.
Experimental Protocols and Implementation
Successful implementation of evaluation frameworks requires detailed experimental protocols and appropriate tool selection. This section provides specific methodologies for key experiments cited throughout this guide.
Protocol for Low-Data Regime Modeling
The ROBERT workflow provides a standardized protocol for machine learning in data-limited chemical applications [62]:
- Data Preparation: Reserve 20% of initial data (minimum four points) as an external test set using "even" distribution to ensure balanced representation
- Hyperparameter Optimization: Employ Bayesian optimization with a combined RMSE objective function incorporating both interpolation (10× 5-fold CV) and extrapolation (selective sorted 5-fold CV) performance
- Model Training: Apply regularization techniques appropriate to each algorithm (L2 for linear models, dropout for neural networks, depth limits for tree-based methods)
- Validation: Execute 10× 5-fold cross-validation to mitigate splitting effects and human bias
- Scoring: Calculate the comprehensive ROBERT score based on predictive ability, overfitting, uncertainty, and robustness checks
Protocol for Uncertainty Quantification
The area metric decomposition protocol enables detailed assessment of probabilistic models [64]:
- Distribution Estimation: For each prediction, estimate full probability distributions using appropriate methods (Bayesian networks, ensemble methods, or direct uncertainty quantification)
- Area Metric Calculation: Compute the area between cumulative distribution functions of predictions and observations across all test cases
- Component Separation: Decompose the area metric into absolute bias (accuracy component) and uncertainty width (precision component)
- Benchmarking: Compare decomposition results against baseline models and established methodological standards
- Visualization: Create reliability diagrams comparing predicted confidence intervals against empirical error rates
Protocol for Reaction Pathway Validation
The ARplorer framework implements a systematic protocol for reaction mechanism validation [4]:
- Active Site Identification: Use Pybel to compile active atom pairs and potential bond-breaking locations
- Structure Optimization: Employ iterative transition state searches combining active-learning sampling with potential energy assessments
- IRC Analysis: Perform intrinsic reaction coordinate calculations to derive new reaction pathways
- Pathway Filtering: Apply general and system-specific chemical logic encoded as SMARTS patterns to eliminate unlikely pathways
- Energy Verification: Execute higher-level theory single-point energy calculations on key stationary points to validate predictions
Table 3: Essential Research Reagent Solutions for Computational Evaluation
| Tool Category | Specific Tools | Primary Function | Application Context |
|---|---|---|---|
| Automated Workflows | ROBERT | Automated ML model development with hyperparameter optimization and evaluation | Low-data regime chemical modeling [62] |
| Pathway Exploration | ARplorer | Automated reaction pathway exploration with QM and rule-based methods | Reaction mechanism studies [4] |
| Uncertainty Quantification | Gaussian Process Models (GPR, GPAR) | Probabilistic prediction with inherent uncertainty estimation | Chemical source term prediction, uncertainty-aware modeling [65] [64] |
| Selectivity Prediction | RegioSQM, RegioML, pKalculator | Site- and regioselectivity prediction for organic reactions | Synthesis planning, reaction outcome prediction [66] |
| Benchmark Datasets | Curated chemical datasets (A-H in ROBERT study) | Standardized performance comparison across methods | Method benchmarking, validation studies [62] |
Model Selection Guide: This decision diagram illustrates the selection of computational methods based on data availability, accuracy requirements, and computational constraints.
The rigorous assessment of predictive accuracy, computational cost, and validity rates forms the foundation of trustworthy computational research in chemical reaction mechanisms. As the field continues to evolve with increasingly sophisticated machine learning methods and automated workflows, standardized evaluation practices become ever more critical. The frameworks, metrics, and protocols presented in this guide provide researchers with comprehensive tools for method validation and comparison. By adopting these structured assessment approaches, the scientific community can accelerate the development of more reliable, efficient, and chemically meaningful computational models that effectively bridge theoretical prediction with experimental reality, ultimately driving innovation across drug discovery, materials science, and catalyst design.
The computational exploration of chemical reaction mechanisms is witnessing a paradigm shift, moving from a competition between physics-based and machine learning (ML) approaches to a strategic integration of both. Physics-based models, grounded in quantum mechanics and molecular mechanics, provide fundamental understanding and reliability but often at a high computational cost. Machine learning models offer unparalleled speed and data-driven pattern recognition but can struggle with generalizability and physical realism. This whitepaper demonstrates that hybrid methodologies are emerging as the superior framework, leveraging the strengths of each approach to deliver accurate, interpretable, and efficient predictions for chemical research and drug development.
Accurately predicting the outcomes and pathways of chemical reactions is a cornerstone of research in fields ranging from medicinal chemistry to materials science. For decades, density functional theory (DFT) and other physics-based simulations have been the workhorses for modeling reaction mechanisms, offering insights grounded in first principles [67]. However, the computational expense of these methods often renders them prohibitive for large-scale screening.
The rise of artificial intelligence has introduced data-driven machine learning models as a powerful alternative. These models, particularly deep learning architectures, can predict reaction outcomes in milliseconds, but their effectiveness is contingent on the quality and breadth of their training data, and they can sometimes produce physically implausible results [8] [68]. This document provides a head-to-head comparison of these philosophies, examining their performance, limitations, and the emerging synergy that is defining the future of the field.
Performance Comparison: A Quantitative Analysis
The table below summarizes the core characteristics, strengths, and weaknesses of physics-based, machine learning, and hybrid approaches.
Table 1: Core Characteristics of Computational Approaches for Reaction Prediction
| Feature | Physics-Based Models | Machine Learning Models | Hybrid Models |
|---|---|---|---|
| Theoretical Basis | First principles (e.g., quantum mechanics) | Statistical patterns in data | Integration of physical laws with data-driven learning |
| Computational Cost | High (hours to days per calculation) | Very low (milliseconds after training) | Moderate (depends on physical model complexity) |
| Data Requirements | Low (theoretical parameters) | Very High (thousands to millions of data points) | Moderate (can work with smaller datasets) |
| Interpretability | High (mechanisms are explicit) | Low ("black box" predictions) | Moderate to High (physical constraints provide insight) |
| Handling of Novelty | Strong (principles are general) | Weak (limited to chemical space of training data) | Strong (guided by principles, refined by data) |
| Key Limitation | Computational cost and scaling | Physical plausibility and data bias | Design complexity and integration overhead |
A direct performance comparison reveals a nuanced picture. For instance, a hybrid model developed for nucleophilic aromatic substitution (SNAr) reactions was trained on only 100-150 experimental rate constants. It achieved a mean absolute error of 0.77 kcal mol⁻¹ on an external test set—surpassing "chemical accuracy" (1 kcal mol⁻¹)—and achieved a top-1 accuracy of 86% on patent reaction data for a task it was not explicitly trained for (regio- and chemoselectivity prediction) [67].
In contrast, purely data-driven models like the Molecular Transformer, while achieving high top-1 accuracy on standardized benchmarks, have been shown to suffer from dataset bias and a lack of interpretability. When evaluated on a debiased dataset, its performance decreased significantly, highlighting that its high accuracy was partly due to learning superficial statistical patterns in the data rather than underlying chemistry [68].
Experimental Protocols & Methodologies
A Pure Machine Learning Workflow: The Molecular Transformer
The Molecular Transformer represents a state-of-the-art pure ML approach, treating reaction prediction as a language translation task.
Table 2: Key Research Reagents for ML-Based Reaction Prediction
| Research Reagent | Function in the Workflow |
|---|---|
| USPTO Dataset | A massive dataset of reactions text-mined from U.S. patents; serves as the primary training corpus [68]. |
| SMILES Strings | Simplified Molecular-Input Line-Entry System; a text-based representation of chemical structures used as the model's input and output [68]. |
| Transformer Architecture | A neural network architecture using self-attention mechanisms to model long-range dependencies in sequence data, originally designed for machine translation [68]. |
Detailed Protocol:
- Data Preparation: A large dataset of reactions (e.g., the USPTO dataset with millions of examples) is curated. Reactants, reagents, and products are converted into SMILES strings.
- Data Augmentation: The dataset is augmented by generating multiple, equivalent SMILES representations for each molecule to improve model robustness [68].
- Model Training: The Transformer model is trained to "translate" a string representing reactants and reagents into a string representing the major product.
- Prediction & Interpretation: For a new set of reactants, the model generates a product SMILES string and a probability score. Interpretation techniques like Integrated Gradients can be used to attribute the prediction to specific parts of the input molecules, and latent space similarity can identify the most influential training examples [68].
A Hybrid Workflow: Flow Matching for Electron Redistribution (FlowER)
The FlowER model, developed at MIT, exemplifies the hybrid approach by tightly integrating physical constraints into a generative AI framework.
Detailed Protocol:
- Physical Representation: Reactions are represented using a bond-electron matrix, a method developed by Ivar Ugi in the 1970s. This matrix explicitly tracks all atoms, bonds, and lone electron pairs, ensuring a physically valid state [8].
- Model Architecture & Training: A generative flow matching model is trained to predict the progression of the reaction mechanism. The model learns to transform the reactant matrix into the product matrix through a series of physically plausible intermediate steps.
- Physical Constraint Enforcement: The bond-electron matrix formalism inherently conserves mass and electrons throughout the generated mechanism. This prevents the model from "creating" or "destroying" atoms, a known failure mode of some LLM-based approaches [8].
- Validation: The model was trained on over a million reactions from a patent database and demonstrated performance matching or exceeding existing approaches while guaranteeing physical validity [8].
A Physics-Consistent ML Workflow: Output Projection onto Physical Manifolds
This methodology ensures physical compliance as a post-processing step, making it model-agnostic.
Detailed Protocol:
- Base Model Prediction: Any ML model (e.g., a neural network) makes an initial prediction for a physical system.
- Constraint Definition: The relevant physical conservation laws (e.g., energy, charge, mass) are defined as a set of constraint equations,
g(x, p) = 0. - Optimization-Based Projection: The initial ML prediction is projected onto the "physical manifold" by solving a constrained optimization problem that finds the closest point to the prediction that satisfies all physical laws. This is formulated as:
- Minimize
|| p - f(x; Θ) ||²with respect top - Subject to
g(x, p) = 0wheref(x; Θ)is the ML model's output andpis the physics-consistent projected output [69].
- Minimize
- Result: This method has been shown to reduce errors in physical law compliance by over four orders of magnitude and improve state variable predictions by up to 72% in benchmark systems like spring-mass oscillators [69].
Visualizing the Hybrid Workflow
The following diagram illustrates the integrated workflow of a hybrid physics-ML model, such as FlowER, showcasing how physical principles are embedded within the AI's architecture.
Diagram 1: Hybrid physics-ML model workflow.
The Scientist's Toolkit: Essential Research Reagents
This table details key computational tools and resources essential for conducting research in this domain.
Table 3: Essential Research Reagents for Computational Reaction Prediction
| Category | Item | Function & Application |
|---|---|---|
| Software & Platforms | AutoDock/SwissADME | Used for in silico screening; predicts binding potential and drug-likeness [38]. |
| CETSA (Cellular Thermal Shift Assay) | Not a computational tool, but a critical experimental method for validating direct target engagement in intact cells, providing ground truth data [38]. | |
| AlphaFold2/3 | Generates highly accurate 3D protein structures from amino acid sequences, expanding the scope of target-centric and physics-based methods [70] [71]. | |
| Datasets & Databases | ChEMBL | A manually curated database of bioactive molecules with drug-like properties, containing quantitative binding data and targets. Essential for training ligand-centric prediction models [70]. |
| USPTO Dataset | A large-scale dataset of organic reactions extracted from U.S. patents, commonly used for training product-prediction models like the Molecular Transformer [68]. | |
| Computational Descriptors | Bond-Electron Matrix | A physics-grounded representation of a reaction that explicitly accounts for atoms, bonds, and electrons, enabling strict adherence to conservation laws [8]. |
| Reaction Fingerprints | Structural or descriptor-based representations (e.g., Morgan fingerprints) that encode chemical reactions for similarity analysis and machine learning [70]. |
The head-to-head comparison conclusively shows that no single approach is universally superior. The choice between physics-based and machine learning methods depends critically on the specific research objective, available data, and required level of interpretability.
However, the most powerful and promising trend is the move toward tightly integrated hybrid models. By embedding physical principles like mass conservation and transition state theory directly into machine learning architectures—as seen with FlowER and physics-consistent projection methods—researchers can create systems that are both data-efficient and physically realistic. The future of computational reaction mechanism exploration lies in this synergistic partnership, which promises to accelerate the discovery of new chemical reactions and optimize synthetic routes for next-generation therapeutics and materials.
The computational exploration of chemical reaction mechanisms represents a cornerstone of modern research in catalysis, drug discovery, and materials science. In 2025, researchers face a critical strategic decision: selecting between powerful commercial platforms and flexible open-source tools to build their computational infrastructure. This choice profoundly impacts not only immediate research capabilities but also long-term innovation velocity, cost structures, and methodological reproducibility. Commercial suites like Schrödinger's platform offer integrated, validated workflows supported by extensive scientific teams, while open-source initiatives provide transparency, customization depth, and avoidance of vendor lock-in. The emergence of sophisticated AI models, including both proprietary systems like GitHub Copilot and open-source alternatives, has further complicated this landscape by introducing powerful assistants for code generation, data analysis, and hypothesis generation. This technical guide provides a comprehensive framework for evaluating these platforms within the specific context of chemical reaction mechanism research, offering data-driven comparisons, implementation protocols, and strategic considerations for research teams operating in both academic and industrial settings.
The Current Software Landscape in Computational Chemistry
Commercial Platform Capabilities: The Schrödinger Ecosystem
Schrödinger's 2025 software releases (2025-3 and 2025-4) demonstrate the integrated, end-to-end capabilities characteristic of mature commercial platforms. Their suite provides specialized tools spanning the entire reaction discovery and optimization pipeline, with several notable advancements for reaction mechanism research [72] [73] [74].
The Life Science Suite now includes Flow matching for Electron Redistribution (FlowER) for predicting reaction outcomes while explicitly conserving mass and electrons—a critical advancement for realistic mechanistic modeling [8]. This approach addresses fundamental physical constraints that often challenge pure machine learning models. The platform also features enhanced Mixed Solvent MD (MxMD) with immiscible probes for identifying cryptic binding pockets and improved Free Energy Perturbation (FEP+) protocols for more accurate binding affinity predictions [72] [74].
For Materials Science, the 2025 releases include automated coarse-grained mapping for proteins, predictive ionic conductivity solutions, and expanded support for machine learning force fields (MLFF), including Universal Models for Atoms (UMA) developed by Meta Platforms Inc. [72] [74]. These capabilities enable researchers to simulate complex systems across multiple time and length scales, from electronic structure calculations to mesoscale phenomena.
The Maestro Graphical Interface serves as the unified environment tying these capabilities together, with the newly introduced AI-powered Maestro Assistant (in beta) providing a conversational interface for querying documentation and controlling the workspace using natural language [73]. This reduces the learning curve for complex simulation setups and enhances researcher productivity.
Open-Source Innovations and Community-Driven Tools
The open-source ecosystem provides compelling alternatives across the computational chemistry workflow, with particular strengths in transparency, modularity, and cost efficiency. Unlike integrated commercial suites, open-source tools often excel at specific tasks and can be combined into customized pipelines.
For reaction prediction and mechanism exploration, MIT's recently developed FlowER (Flow matching for Electron Redistribution) represents a significant open-source advancement. This generative AI approach uses a bond-electron matrix based on 1970s work by Ivar Ugi to explicitly track all electrons in a reaction, ensuring conservation of mass and electrons while predicting outcomes [8]. The model was trained on over a million chemical reactions from the U.S. Patent Office database and is available open-source on GitHub, providing researchers with full transparency into the methodology and the ability to customize the implementation for specific reaction classes [8].
For general process simulation, DWSIM stands out as a fully open-source chemical process simulator that offers comprehensive capabilities for modeling various unit operations, including reactors and distillation columns. Its Python integration enables extensive customization and automation, making it particularly valuable for research applications requiring modification of underlying algorithms [75].
The broader AI-assisted development landscape is dominated by GitHub Copilot, which has seen remarkable adoption with over 15 million users globally by 2025, including 90% of Fortune 100 companies [76]. While not specific to computational chemistry, its productivity benefits for researchers developing custom analysis scripts, workflow automations, and simulation extensions are substantial. Studies indicate developers code 51% faster using Copilot, with an 84% increase in successful builds in enterprise deployments [76].
Table 1: Quantitative Comparison of Representative Platforms for Reaction Mechanism Research
| Platform | Licensing Model | Key Features for Reaction Research | Typical Deployment Scope | Primary Strengths |
|---|---|---|---|---|
| Schrödinger Suite | Commercial | FEP+, MxMD, FlowER, MLFF support [72] [74] | Enterprise-wide integration | Validated workflows, integrated UI, professional support |
| FlowER (MIT) | Open-Source | Electron-redistribution reaction prediction [8] | Specific research projects | Transparency, physical constraints, customization |
| DWSIM | Open-Source | Process simulation, thermodynamic calculations [75] | Educational & specialized professional use | Zero cost, Python extensibility, active community |
| GitHub Copilot | Commercial/Subscription | AI-powered code completion [76] | Individual developers & teams | 51% faster coding, 88% code retention [76] |
Quantitative Cost-Benefit Analysis
Total Cost of Ownership Framework
Evaluating software platforms requires looking beyond initial licensing fees to consider the total cost of ownership (TCO), which includes implementation, training, maintenance, and scaling expenses. For commercial platforms, while subscription costs are transparent, organizations must factor in annual maintenance fees (typically 18-25% of initial license cost), training requirements for complex interfaces, and potential vendor lock-in consequences [77].
Open-source software eliminates licensing fees but introduces other cost considerations. Organizations typically spend an average of 14% more on specialized IT talent for managing open-source implementations, with mid-size enterprises reporting annual expenditures of $40,000-$90,000 on customization and integration [77]. However, this investment often yields significant long-term dividends through customized workflows that precisely match research needs.
Table 2: Cost Structure Analysis for Software Platforms in Research Environments
| Cost Factor | Commercial Platforms (e.g., Schrödinger) | Open-Source Platforms (e.g., DWSIM, FlowER) |
|---|---|---|
| Initial Acquisition | Substantial licensing fees; subscription models [77] | Free licensing; possible implementation consulting costs [75] |
| Maintenance & Support | Annual fees (18-25% of license); included support [77] | Internal staff costs; commercial support contracts optional [77] |
| Customization | Vendor-dependent; often limited to supported features | Complete flexibility; costs depend on internal expertise [78] |
| Training | Vendor-provided training; standardized materials | Community-supported documentation; potentially steeper learning curve |
| Scalability | Per-user or per-core licensing; predictable cost scaling | Infrastructure costs only; highly scalable with appropriate expertise |
| Vendor Lock-in Risk | High (proprietary formats, workflow dependency) [77] | Minimal (open standards, extensible code) |
| 5-Year TCO Projection | Higher initial, predictable annual costs | Lower initial, variable based on internal support needs [78] |
Productivity and Output Metrics
Quantifying the impact of software platforms on research productivity provides crucial insights for decision-making. GitHub's research indicates that developers using Copilot code 51% faster for certain tasks, with an 8.69% increase in pull requests per developer and an 11% increase in pull request merge rates [76]. Perhaps most significantly, Copilot users experience an 84% increase in successful builds, suggesting higher initial code quality [76].
For scientific platforms specifically, Schrödinger's continuous interface improvements and workflow automation aim to reduce the time from experimental design to simulation results. Features like the FEP Protocol Builder's automated machine learning workflow can generate optimized models up to two times faster, while Optimized Glide enables screening large compound libraries in roughly half the previous compute time [73].
A balanced approach often yields optimal results. Case studies show that organizations implementing hybrid models—using open-source cores with commercial extensions or support—can reduce five-year TCO by approximately 42% while maintaining capabilities and compliance [77].
Implementation Protocols for Reaction Mechanism Research
Workflow for Comparative Reaction Barrier Analysis
A critical application in computational reaction research involves comparing energy barriers across different catalytic systems or reaction conditions. The following protocol outlines a standardized approach applicable across platforms:
Step 1: System Preparation
- Commercial Platform: Use Maestro's Protein Preparation Wizard for biomolecular systems or the Materials Science Builder for heterogeneous catalysts. Apply standardized optimization protocols with OPLS4 force field.
- Open-Source Alternative: Use Open Babel for initial structure conversion and RDKit for molecular standardization. Apply geometric optimization with UFF or MMFF94s through Python scripts.
Step 2: Transition State Search
- Commercial Platform: Utilize Jaguar's transition state optimization with automated follow-up frequency calculations to confirm saddle points.
- Open-Source Alternative: Implement transition state search using the Growing String Method or similar approaches within open-source packages like ASE (Atomic Simulation Environment) with ORCA as the quantum chemistry backend.
Step 3: Energy Refinement
- Commercial Platform: Execute single-point energy calculations at the DFT level (e.g., B3LYP-D3/6-311+G) using Jaguar's optimized workflows.
- Open-Source Alternative: Run similar calculations using open-source DFT codes like Psi4 or GPAW with consistent functional and basis set selections.
Step 4: Data Analysis and Visualization
- Commercial Platform: Use Maestro's analysis panels for automated reaction coordinate plotting and energy comparison.
- Open-Source Alternative: Develop custom Python scripts using Matplotlib or Plotly for visualization, leveraging GitHub Copilot to accelerate plotting code generation [76].
Diagram 1: Reaction mechanism analysis workflow showing platform decision points.
Research Reagent Solutions: Computational Tools Inventory
Table 3: Essential Computational Research Reagents for Reaction Mechanism Studies
| Tool/Category | Function in Research | Example Platforms | Typical Application Context |
|---|---|---|---|
| Quantum Chemistry Packages | Electronic structure calculation for accurate energy predictions | Jaguar (Commercial), Psi4 (Open) [74] [75] | Reaction barrier calculation, spectroscopic property prediction |
| Molecular Dynamics Engines | Sampling configurational space and time-dependent phenomena | Desmond (Commercial), GROMACS (Open) [74] | Solvation effects, conformational changes, binding pathways |
| Reaction Prediction AI | Predicting reaction products and plausible mechanisms | FlowER (Open) [8] | Hypothesis generation, retrosynthetic analysis, mechanism proposal |
| Process Simulators | Modeling larger-scale chemical processes with multiple unit operations | Aspen HYSYS (Commercial), DWSIM (Open) [75] | Scale-up considerations, process optimization, cost analysis |
| Code Completion AI | Accelerating custom script and analysis pipeline development | GitHub Copilot (Commercial) [76] | Data processing, visualization, workflow automation |
| Force Field Libraries | Providing parameters for classical molecular mechanics simulations | OPLS4 (Commercial), Open Force Field (Open) [74] | Large system simulations, initial structure optimization |
Strategic Implementation Framework
Organizational Capability Assessment
Selecting between open-source and commercial platforms requires honest assessment of organizational capabilities across several dimensions:
Technical Expertise: Commercial platforms like Schrödinger provide intuitive interfaces that reduce the expertise barrier for complex simulations, while open-source tools typically require deeper computational chemistry and programming knowledge. Organizations with strong computational chemistry groups possessing programming skills (Python, C++) can leverage open-source tools more effectively, while those relying on researchers primarily trained in experimental techniques may benefit from commercial platforms' guided workflows.
IT Infrastructure and Support: Commercial vendors manage software updates, compatibility, and technical support, reducing internal IT burdens. Open-source solutions require internal maintenance, updating, and troubleshooting capabilities, though this can be offset by the flexibility to fix issues directly without vendor dependency.
Research Flexibility vs. Standardization Needs: Open-source platforms enable complete customization of methods and algorithms—critical for developing novel computational approaches. Commercial platforms offer standardized, validated protocols that enhance reproducibility and are often preferred in regulated environments like pharmaceutical development.
Diagram 2: Strategic decision framework for platform selection.
Hybrid Implementation Model
Most research organizations benefit from a hybrid approach that strategically combines both platform types:
Core Infrastructure with Commercial Platforms: Utilize commercial software for standardized, validated calculations requiring reproducibility and support, such as FEP+ calculations for binding affinity predictions or polymorph predictions for formulation development [72] [74].
Innovation and Customization with Open-Source: Employ open-source tools for novel method development, specialized analyses not supported in commercial packages, and educational purposes where transparency is essential.
AI-Assisted Productivity Across Environments: Implement GitHub Copilot or similar tools to enhance productivity across all computational work, particularly for developing custom analysis scripts, automating workflows, and generating documentation [76].
A biotech case study demonstrated the effectiveness of this approach, where a team combined Schrödinger's FEP+ for binding affinity predictions with custom open-source analysis scripts (developed more rapidly using GitHub Copilot) to accelerate preclinical candidate identification from years to months [79].
The choice between open-source and commercial platforms for computational reaction mechanism research is not binary but strategic. Commercial platforms like Schrödinger offer robust, validated, and integrated environments that reduce time-to-solution for standard problems, while open-source tools provide transparency, customizability, and avoidance of vendor lock-in. The emerging generation of AI-assisted tools, exemplified by GitHub Copilot, is becoming an essential layer across both environments, significantly enhancing researcher productivity.
Future developments will likely further blur the boundaries between these paradigms, with commercial platforms incorporating more open-source components and open-source projects developing commercial support models. The most successful research organizations will be those that develop the strategic capability to evaluate and implement the right combination of tools for their specific research challenges, technical capabilities, and budgetary constraints. By taking a hybrid, purpose-driven approach to platform selection, research teams can maximize both productivity and innovation in their computational exploration of chemical reaction mechanisms.
Within the broader thesis of computational exploration of chemical reaction mechanisms, the ability to not only predict but also to provide experimentally validated insights represents a significant frontier. For decades, computational chemistry has provided critical insights, but a key challenge has been grounding these predictions in real-world physical constraints to ensure their reliability and applicability in fields like drug development [80]. Early models often overlooked finer details of electron movements and reactive intermediates, leading to a disconnect between computational forecasts and experimentally observable outcomes [81]. This review details a transformative shift, highlighting contemporary case studies where advanced computational approaches, integrating physical laws and large-scale data, have generated predictions that stand up to rigorous experimental validation, thereby providing researchers with powerful new tools for synthetic planning.
Case Study: FlowER - Physically Constrained Prediction of Reaction Outcomes
Experimental Protocol and Methodology
A team at MIT addressed a fundamental limitation in previous AI-driven reaction prediction models: their frequent violation of basic physical principles like the conservation of mass [8]. Their model, FlowER (Flow matching for Electron Redistribution), introduces a novel methodology to incorporate these constraints.
- Core Computational Framework: FlowER utilizes a bond-electron matrix, a method originally developed in the 1970s by chemist Ivar Ugi, to represent the electrons involved in a reaction [8]. This matrix uses nonzero values to represent bonds or lone electron pairs and zeros to represent their absence, providing a foundational structure that inherently respects conservation laws.
- Training and Validation Data: The model was trained on a large-scale dataset comprising over a million chemical reactions obtained from a U.S. Patent Office database [8]. This grounds the model in experimentally validated data from the patent literature, ensuring the inferred mechanisms are not merely theoretical but are imputed from real-world results.
- Mechanism Inference: Instead of merely mapping inputs to outputs, the system is designed to track all chemicals and how they are transformed throughout the entire reaction process. This allows for the explicit conservation of both atoms and electrons, preventing the model from "creating" or "deleting" atoms, a pitfall of previous LLM-based approaches [8].
Significance and Validation
The development of FlowER represents a proof of concept that generative AI and flow matching are exceptionally well-suited for chemical reaction prediction [8]. The model matches or outperforms existing approaches in finding standard mechanistic pathways while ensuring near-perfect validity and conservation. This reliability makes it a valuable tool for researchers assessing reactivity and mapping out reaction pathways, with potential applications in medicinal chemistry and materials discovery [8]. Its open-source availability on GitHub ensures broad accessibility for the scientific community, acting as a stepping stone toward inventing new reactions and advancing mechanistic understanding [8].
Case Study: ARplorer - Automated Exploration of Reaction Pathways
Experimental Protocol and Methodology
The ARplorer program addresses the challenge of efficiently exploring complex potential energy surfaces (PES) for multi-step reactions, a task that is notoriously time-consuming for conventional quantum mechanics (QM) and molecular dynamics (MD) simulations [4]. ARplorer integrates multiple advanced computational techniques into a cohesive, automated workflow.
- Algorithmic Workflow: ARplorer operates on a recursive algorithm where each iteration involves several steps. The program first identifies active sites and potential bond-breaking locations to set up input molecular structures. It then optimizes molecular structures through iterative transition state (TS) searches, employing a blend of active-learning sampling and potential energy assessments. Finally, it performs Intrinsic Reaction Coordinate (IRC) analysis to derive new reaction pathways, eliminates duplicates, and finalizes the structure for the next input [4].
- Multi-Level Computational Strategy: For efficiency and accuracy, ARplorer combines the semi-empirical GFN2-xTB method for quick PES generation with Gaussian 09's algorithm for TS searching. This allows for flexibility, enabling researchers to use GFN2-xTB for large-scale screening and more precise Density Functional Theory (DFT) for detailed calculations [4].
- LLM-Guided Chemical Logic: A key innovation is the integration of a Large Language Model (LLM) to assist in building chemical logic. This logic is derived from two sources: a pre-generated knowledge base from literature (books, databases, research articles) and system-specific rules generated by querying specialized LLMs with the reaction system in SMILES format. This guides the PES exploration by filtering unlikely pathways and focusing the search on chemically plausible routes [4].
Significance and Validation
ARplorer's effectiveness and versatility have been demonstrated through case studies on complex multi-step reactions, including organic cycloaddition, asymmetric Mannich-type reactions, and organometallic Pt-catalyzed reactions [4]. By integrating rule-based practicality with precise QM techniques and literature-derived chemical logic, ARplorer achieves accurate and efficient identification of reaction pathways, significantly improving computational efficiency over conventional approaches. Its capability for high-throughput screening positions it as an efficient tool for data-driven reaction development and catalyst design [4].
Quantitative Performance Comparison of Computational Methods
The table below summarizes the key quantitative and methodological aspects of the featured computational approaches, allowing for direct comparison of their capabilities and foundations.
Table 1: Comparative Analysis of Experimentally Validated Computational Prediction Methods
| Method / Model | Core Approach | Training/Validation Data | Key Performance Metrics | Experimental Validation & Applications |
|---|---|---|---|---|
| FlowER [8] | Generative AI with physical constraints (bond-electron matrix). | >1 million reactions from USPTO database. | Massive increase in validity/conservation; matching or better accuracy vs. existing models. | Open-source tool for assessing reactivity & mapping pathways; applications in medicinal chemistry & materials discovery. |
| ARplorer [4] | QM + rule-based PES exploration with LLM-guided chemical logic. | Case studies (e.g., cycloaddition, Mannich-type, Pt-catalyzed reactions). | Significant improvements in computational efficiency & practicality vs. conventional QM/MD. | Efficient tool for data-driven reaction development & catalyst design; validated via case studies. |
| MechFinder [81] | Automated mechanistic labeling using reaction & mechanistic templates. | USPTO-33K dataset (subset of USPTO-50K). | Generates first large-scale dataset (mech-USPTO-31K) of chemically reasonable mechanisms. | Dataset provides benchmark for developing mechanism-based prediction models. |
Experimental Protocols for Mechanistic Template Generation
The creation of large-scale, reliable datasets of reaction mechanisms is a critical prerequisite for training robust predictive models. The MechFinder method provides a detailed protocol for this purpose [81].
- Reaction Template (RT) Extraction: The process begins by identifying "changed atoms" through a comparison of the chemical environment of each atom before and after a reaction, guided by atom-mapping. The scope of the template is then extended to include neighboring atoms connected by double, triple, or aromatic bonds, as well as atoms within manually defined mechanistically important special groups (e.g., carbonyl groups). Finally, chemical fragments from both reactants and products are extracted in SMARTS format based on the identified atoms and connected to form the reaction template [81].
- Mechanistic Template (MT) Hand-Coding: For a given group of reactions (Mechanistic Class, or MC) that follow the same mechanism, an MT is hand-coded. This MT describes the sequence of arrow-pushing diagrams, representing attacking and electron-receiving moieties. The MTs are designed to distinguish between different mechanisms that share the same RT (e.g., SN1 vs. SN2) using chemically designed criteria. This step also involves recovering necessary reagents that are missing from the original reaction data but are essential for the mechanism to proceed [81].
Visualizing Automated Reaction Pathway Exploration
The following diagram illustrates the integrated computational workflow of the ARplorer program, which combines quantum mechanics, active learning, and chemical logic to automate the exploration of reaction pathways.
Visualizing Mechanistic Dataset Curation
The mech-USPTO-31K dataset provides a critical benchmark for developing mechanistic prediction models. The workflow below details the process of its creation.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key computational and data resources that are essential for conducting research in computationally predicted reaction mechanisms.
Table 2: Key Research Reagent Solutions for Computational Reaction Prediction
| Tool / Resource | Type | Primary Function in Research |
|---|---|---|
| Bond-Electron Matrix [8] | Computational Representation | Foundational data structure for representing electrons and bonds, ensuring conservation of mass and electrons in reaction prediction models. |
| mech-USPTO-31K Dataset [81] | Benchmarking Dataset | A large-scale dataset of chemically reasonable reaction mechanisms with arrow-pushing diagrams, used to train and validate new prediction models. |
| Reaction Template (RT) [81] | Data Extraction Method | Automatically extracted rule capturing the localized chemical transformation in a reaction, focusing on changed and extended atoms. |
| Mechanistic Template (MT) [81] | Expert-Coded Rule | Hand-coded sequence of arrow-pushing diagrams that describe the electron movements for a specific class of reactions, providing ground-truth mechanisms. |
| GFN2-xTB [4] | Computational Method | Semi-empirical quantum mechanical method used for rapid generation of potential energy surfaces and large-scale screening in automated pathway exploration. |
Conclusion
The integration of AI with foundational physical principles is fundamentally reshaping the exploration of chemical reaction mechanisms. The synergy between generative models like FlowER, LLM-guided explorers like ARplorer, and robust quantum methods has created an unprecedented capability to predict pathways and transition states with high accuracy while respecting real-world constraints. These computational advances are dramatically streamlining drug discovery, as evidenced by the ability to screen billions of compounds and identify clinical candidates in months rather than years. Future progress hinges on expanding these models to encompass a broader range of chemistries, particularly organometallics and catalysis, and on deeper integration of active learning for autonomous discovery. For biomedical research, this computational transformation promises not only faster development of safer, more effective drugs but also the democratization of discovery, empowering researchers to tackle increasingly complex therapeutic challenges.
References
- 1. Accelerating compound synthesis in drug discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560346/]
- 2. Quantitative interpretation explains machine learning models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7966799/]
- 3. A deep learning framework for accurate reaction prediction ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00732-w]
- 4. Large language model guided automated reaction ... [https://www.nature.com/articles/s42004-025-01630-y]
- 5. RxnNet: An AI Framework for Reaction Mechanism Discovery [https://chemrxiv.org/engage/chemrxiv/article-details/690510f6ef936fb4a2a1cfc0]
- 6. Conservation of mass - Wikipedia [https://en.wikipedia.org/wiki/Conservation_of_mass]
- 7. A Nudge to the Truth: Atom Conservation as a Hard Constraint in Models of Atmospheric Composition Using a Species-Weighted Correction - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11730974/]
- 8. A new generative AI approach to predicting chemical ... [https://news.mit.edu/2025/generative-ai-approach-to-predicting-chemical-reactions-0903]
- 9. 4.2: Law of Conservation of Mass - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Introductory_Chemistry/Introductory_Chemistry_(CK-12)/04:_Atomic_Structure/4.02:_Law_of_Conservation_of_Mass]
- 10. Heuristics-Guided Exploration of Reaction Mechanisms [https://arxiv.org/abs/1509.03120]
- 11. A Trajectory-Based Method to Explore Reaction Mechanisms [https://pmc.ncbi.nlm.nih.gov/articles/PMC6321347/]
- 12. Quantum Chemistry in Drug Discovery | Rowan [https://rowansci.com/publications/quantum-chemistry-in-drug-discovery]
- 13. A human-machine interface for automatic exploration of ... [https://www.nature.com/articles/s41467-024-47997-9]
- 14. Electron flow matching for generative reaction mechanism ... [https://arxiv.org/html/2502.12979v1]
- 15. Electron Flow Matching Advances Reaction Mechanism ... [https://bioengineer.org/electron-flow-matching-advances-reaction-mechanism-prediction/]
- 16. MIT's FlowER: The AI That Actually Respects the Laws of ... [https://medium.com/@cvinbusiness/mits-flower-the-ai-that-actually-respects-the-laws-of-chemistry-892c4c60ba10]
- 17. A generative Artificial Intelligence approach to predicting ... [https://epium.com/news/generative-artificial-intelligence-predicting-chemical-reactions/]
- 18. A New Approach to Reaction Prediction in Nature Magazine" [https://www.linkedin.com/posts/connorcoley_a-new-generative-ai-approach-to-predicting-activity-7370827900939411456-J94V]
- 19. Schrödinger equation [https://en.wikipedia.org/wiki/Schr%C3%B6dinger_equation]
- 20. Quantum Chemistry: Understanding the Schrödinger Equation [https://rosemaryfrancis.medium.com/quantum-chemistry-understanding-the-schr%C3%B6dinger-equation-588a9ba0b14a]
- 21. A large-scale reaction dataset of mechanistic pathways ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11316731/]
- 22. Electron flow matching for generative reaction mechanism ... [https://www.nature.com/articles/s41586-025-09426-9]
- 23. Electron flow matching for generative reaction mechanism ... [https://arxiv.org/abs/2502.12979]
- 24. A new generative AI approach to predicting chemical ... [https://www.eecs.mit.edu/a-new-generative-ai-approach-to-predicting-chemical-reactions/]
- 25. FongMunHong/FlowER: Flow Matching for Electron ... [https://github.com/FongMunHong/FlowER]
- 26. ECTS: An ultra-fast diffusion model for exploring chemical ... [https://chemrxiv.org/engage/chemrxiv/article-details/67cf041bfa469535b9bc28d4]
- 27. Atom-anchored LLMs speak Chemistry: A Retrosynthesis ... [https://arxiv.org/html/2510.16590v1]
- 28. Multiscale Methods in Drug Design Bridge Chemical and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6445369/]
- 29. Assessing the accuracy and efficacy of multiscale ... [https://www.nature.com/articles/s41598-024-67468-x]
- 30. From QM/MM to ML/MM: A new era in multiscale modeling [https://chemrxiv.org/engage/chemrxiv/article-details/6894368d728bf9025e421b0d]
- 31. Accurate Free Energy Calculation via Multiscale Simulations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288004/]
- 32. Machine learning potential-accelerated multiscale ... [https://pubmed.ncbi.nlm.nih.gov/41133880/]
- 33. Lead Optimization Services in Drug Discovery [https://www.archivemarketresearch.com/reports/lead-optimization-services-in-drug-discovery-141080]
- 34. How Machine Learning is Recoding the Future of Drug ... [https://www.drugpatentwatch.com/blog/the-application-of-machine-learning-in-drug-discovery-revolutionizing-pharmaceutical-research/?srsltid=AfmBOooIL_vSVA7iel_4f5qX5TKrLJeDOm4yy04S0-oDDcJ3yWaEAk-z]
- 35. Accelerating Drug Development with AI in the U.S. ... [https://intuitionlabs.ai/articles/accelerating-drug-development-ai-pharma]
- 36. AI-Based Pharmaceutical R&D Services Market on a [https://www.globenewswire.com/news-release/2025/11/20/3192101/0/en/AI-Based-Pharmaceutical-R-D-Services-Market-on-a-High-Growth-Trajectory-report-by-Towards-Healthcare.html]
- 37. Keynote: Accelerating Drug Discovery and Development with ... [https://aaps2025.eventscribe.net/ajaxcalls/PresentationInfo.asp?PresentationID=1592519]
- 38. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 39. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 40. Future-proofing pharma R&D labs | Deloitte Insights [https://www.deloitte.com/us/en/insights/industry/health-care/future-proofing-pharma-rnd-labs.html]
- 41. Predicting reaction conditions from limited data through active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9172577/]
- 42. Transfer learning across different chemical domains: virtual ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00886-1]
- 43. Application of the digital annealer unit in optimizing chemical ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01043-y]
- 44. Data Augmentation and Transfer Learning Strategies for ... [https://chemrxiv.org/engage/chemrxiv/article-details/60c75312469df47cd5f44ca9]
- 45. Transfer learning from custom-tailored virtual molecular ... [https://www.nature.com/articles/s42004-025-01678-w]
- 46. ReactionT5: a pre-trained transformer model for accurate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12366004/]
- 47. Highly parallel optimisation of chemical reactions through ... [https://www.nature.com/articles/s41467-025-61803-0]
- 48. Prediction of energy consumption in four sectors using ... [https://www.sciencedirect.com/science/article/pii/S2405844025001458]
- 49. Quantum Grid Path Planning Using Parallel QAOA Circuits ... [https://arxiv.org/html/2510.07413v1]
- 50. Assessing the complexity of a path search optimization ... [https://epjdatascience.springeropen.com/articles/10.1140/epjds/s13688-025-00542-0]
- 51. Reducing complexity in an agent based reaction model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5000584/]
- 52. Catalysis meets machine learning: a guide to data-driven ... [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc05274b?page=search]
- 53. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 54. Precursor reaction pathway leading to BiFeO 3 formation [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00160a]
- 55. Modeling Thia-Michael Reactions - Rowan Scientific [https://www.rowansci.com/blog/modeling-thia-michael-reactions]
- 56. g-xTB - A General-Purpose Tight-Binding Electronic-Structure ... [https://rowansci.com/features/g-xtb]
- 57. Benchmark of Approximate Quantum Chemical and Machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288007/]
- 58. The Case of GFN2-xTB [https://pubmed.ncbi.nlm.nih.gov/37676972/]
- 59. Machine-Learning Adaptivity, DFT-Level Accuracy, and ... [https://chemrxiv.org/engage/chemrxiv/article-details/69004d34113cc7cfff7de942]
- 60. Data-driven discovery of chemical reaction mechanisms ... [https://chemrxiv.org/engage/chemrxiv/article-details/6808813250018ac7c5bb7c11]
- 61. Sparse identification of chemical reaction mechanisms from ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00293a]
- 62. Machine learning workflows beyond linear models in low-data ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00996k]
- 63. Evaluation Metrics for ML Models in Drug Discovery [https://www.elucidata.io/blog/evaluation-metrics-for-ml-models-in-drug-discovery]
- 64. Metrics for Evaluating Machine Learning Models Prediction ... [https://www.sciencedirect.com/science/article/abs/pii/B9780443152740502110]
- 65. Probabilistic machine learning framework for chemical ... [https://www.sciencedirect.com/science/article/pii/S0360319924028945]
- 66. Computational tools for the prediction of site - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc00541h]
- 67. Machine learning meets mechanistic modelling for accurate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9528810/]
- 68. Quantitative interpretation explains machine learning ... [https://www.nature.com/articles/s41467-021-21895-w]
- 69. Physics-consistent machine learning with output projection ... [https://www.nature.com/articles/s42005-025-02329-1]
- 70. A precise comparison of molecular target prediction methods [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00199d]
- 71. Physics-based Modeling in the New Era of Enzyme Engineering [https://pmc.ncbi.nlm.nih.gov/articles/PMC12239909/]
- 72. Explore our latest software - Schrödinger [https://www.schrodinger.com/release-download/]
- 73. Schrödinger releases 2025-3 software with new features ... [https://www.linkedin.com/posts/schr-dinger_schr%C3%B6dinger-continuously-develops-scientific-activity-7358208075494817792-uYVn]
- 74. Schrödinger Release Notes - Release 2025-4 [https://www.schrodinger.com/life-science/download/release-notes/]
- 75. Top 10 Chemical Engineering Tools in 2025 [https://www.devopsschool.com/blog/top-10-chemical-engineering-tools-in-2025-features-pros-cons-comparison/]
- 76. GitHub Copilot Statistics & Adoption Trends [2025] [https://www.secondtalent.com/resources/github-copilot-statistics/]
- 77. Should You Choose Open Source or Proprietary Software ... [https://www.getmonetizely.com/articles/should-you-choose-open-source-or-proprietary-software-a-complete-cost-analysis]
- 78. Open Source vs Proprietary: Save $100K+ Annually in 2025 [https://tech.grahammiranda.com/open-source-proprietary-software-cost-savings-2025/]
- 79. AI Tools Every Scientist Should Know in 2025 [https://www.linkedin.com/pulse/ai-tools-every-scientist-should-know-2025-hossein-maleki-y6uoe]
- 80. A Review of Theoretical Chemical Predictions Ahead ... [https://chemrxiv.org/engage/chemrxiv/article-details/67c0752e6dde43c908b7b7ed]
- 81. A large-scale reaction dataset of mechanistic pathways ... [https://www.nature.com/articles/s41597-024-03709-y]