Benchmarking Bond Energy Accuracy: A Practical Guide to Electronic Structure Methods for Research and Drug Development
Accurately predicting bond dissociation enthalpies (BDEs) and interaction energies is critical for advancing research in catalysis, material science, and rational drug design.
Benchmarking Bond Energy Accuracy: A Practical Guide to Electronic Structure Methods for Research and Drug Development
Abstract
Accurately predicting bond dissociation enthalpies (BDEs) and interaction energies is critical for advancing research in catalysis, material science, and rational drug design. This article provides a comprehensive guide for researchers and drug development professionals, exploring the foundational principles of bond energy calculation, from basic thermodynamic definitions to advanced quantum mechanical models. We survey the current landscape of computational methods—including density functional theory (DFT), semiempirical approaches, and neural network potentials—benchmarking their accuracy and computational cost against experimental data. The content further delivers practical strategies for troubleshooting and optimizing calculations, validates methods through comparative analysis of modern benchmark sets like ExpBDE54, and concludes with actionable insights for selecting the most efficient computational workflow for specific biomedical applications.
The Fundamentals of Bond Energy: From Theoretical Concepts to Experimental Benchmarks
Defining Bond Energy, Enthalpy, and Cohesive Energy in Chemical Systems
In computational chemistry and materials science, accurately predicting the energy associated with forming and breaking bonds is fundamental to understanding stability, reactivity, and properties of molecules and materials. The accuracy of these predictions hinges on the choice of electronic structure method. This guide compares the performance of different computational approaches for determining bond energy, enthalpy, and cohesive energy, providing researchers with a framework for selecting appropriate methodologies.
Bond Energy (or Bond Dissociation Energy, BDE) is defined as the energy required to break a specific chemical bond in a molecule via homolytic cleavage in the gaseous state, resulting in two radical fragments [1] [2]. It is a measure of bond strength; a higher bond energy indicates a stronger, more stable bond. For example, the C-H bond in methane (CH₄) has a bond energy of approximately 104 kcal/mol [2]. Bond energies are correlated with the stability of the resulting radical species; low bond energies often reflect the formation of stable free radicals [2].
Bond Enthalpy is typically used interchangeably with bond energy in many contexts, representing the average energy required to break one mole of a specific type of bond in the gaseous state [1].
Cohesive Energy is the energy required to decompose a solid material or nanocluster into its constituent isolated atoms [3] [4]. It represents the total binding energy of the material and is a crucial parameter for predicting thermodynamic stability. For nanoparticles and nanoclusters, cohesive energy exhibits strong size dependence, decreasing with particle size due to the increased surface-to-volume ratio and quantum confinement effects [3] [4].
Performance Comparison of Electronic Structure Methods
The accuracy of predicting these energy quantities varies significantly across computational methods. The following table summarizes the key characteristics, accuracy, and computational cost of prominent electronic structure techniques.
Table 1: Comparison of Electronic Structure Methods for Energy Calculations
| Method | Theoretical Foundation | Typical Accuracy | Computational Cost | Best Suited For |
|---|---|---|---|---|
| Coupled-Cluster (CCSD(T)) | Gold-standard wavefunction theory [5] | Chemical Accuracy (< 1 kcal/mol) [5] | Very High (O(N⁷)) [5] | Small molecules (tens of atoms) [5] |
| Density Functional Theory (DFT) | Electron density functionals [6] [5] | Variable; can be good but not uniform [5] | Moderate (O(N³)) [5] | Medium/large systems (hundreds of atoms) [5] |
| Machine Learning (e.g., MEHnet) | Trained on high-level data (e.g., CCSD(T)) [5] | Near-CCSD(T) accuracy [5] | Low (after training) | High-throughput screening of large systems [5] |
| Bond Energy Model (BEM) | Empirical model based on bond counting [3] [4] | Lower; depends on parameterization [4] | Very Low | Large nanoparticles for trend analysis [3] |
The performance comparison reveals a direct trade-off between accuracy and computational scalability. CCSD(T) is the undisputed benchmark for accuracy on small systems. DFT provides the best balance of accuracy and system size for many materials science applications, though its performance depends heavily on the chosen exchange-correlation functional [6]. Emerging machine learning potentials, like MEHnet, show promise in bridging this gap by offering CCSD(T)-level accuracy at a fraction of the cost, enabling the study of thousands of atoms [5].
Table 2: Illustrative Bond Energy Data from Different Methodologies
| Bond / System | Experimental / Reference Value | CCSD(T) Prediction | GGA-DFT Prediction | Empirical Model |
|---|---|---|---|---|
| C-H (in CH₄) | 104 kcal/mol [2] | Near exact [5] | ~100-105 kcal/mol (functional dependent) | - |
| H⁺·N₂ Bond Energy | 113.7 kcal/mol [7] | - | - | 141.5 kcal/mol (STO-3G) [7] |
| Cohesive Energy of Pt Nanocluster | - | -2.95 eV/atom (3-layer) [4] | -2.5 eV/atom (Bond Energy Model) [4] | - |
| Band Gap of BaZrO₃ | 4.7-4.9 eV (indirect) [6] | - | ~3.1-3.2 eV (GGA/LDA) [6] | - |
Experimental and Computational Protocols
Protocol for Calculating Cohesive Energy via First-Principles DFT
This protocol is widely used for determining the cohesive energy of nanoclusters and solid-state materials [4].
- Geometry Optimization: Perform a full structural relaxation of the nanoparticle or crystal system using DFT (e.g., VASP, CASTEP) until the forces on atoms are minimized (e.g., below 0.01 eV/Å) [4] [6].
- Total Energy Calculation: Calculate the total energy (E_tot) of the fully optimized system.
- Atomic Energy Calculation: Calculate the energy of an isolated, single constituent atom (E_atom) of the material.
- Cohesive Energy Computation: Calculate the cohesive energy per atom (Ecoh) using the formula: *Ecoh = Eatom - Etot / n* where n is the number of atoms in the system [4].
Protocol for Bond Energy Decomposition Analysis (ADF)
This protocol, as implemented in the ADF software, decomposes the bond energy into chemically meaningful components [8].
- Fragment Definition: Define the molecular system as two or more interacting fragments (e.g., a base and a substrate).
- Preparation Energy: Calculate the energy required (ΔE_prep) to deform the isolated fragments from their equilibrium geometry to the geometry they adopt in the final molecule.
- Interaction Energy Decomposition: Calculate the interaction energy (ΔEint) between the prepared fragments and decompose it as follows:
- Electrostatic Interaction (ΔVelst): The classical attraction between the unperturbed charge distributions of the fragments.
- Pauli Repulsion (ΔEPauli): The destabilizing energy arising from the antisymmetrization of the fragment wavefunctions, responsible for steric repulsion.
- Orbital Interaction (ΔEoi): The energy from electron pair bonding, charge transfer, and polarization [8].
Workflow for High-Throughput Screening with Machine Learning
This modern workflow accelerates the prediction of bond-related energies with high accuracy [5].
- Training Set Generation: Perform CCSD(T) calculations on a diverse set of small molecules (typically up to 10 atoms) to generate a reference dataset.
- Neural Network Training: Train a specialized equivariant graph neural network (e.g., MEHnet) on this dataset. The model learns to map molecular structures to electronic properties.
- Property Prediction: Use the trained model to predict the total energy, cohesive energy, bond energies, and other electronic properties (dipole moment, polarizability, excitation gaps) for new, larger molecules (thousands of atoms) at near-CCSD(T) accuracy and low computational cost [5].
Diagram 1: Method selection workflow for accurate energy prediction.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools for Energy Calculations
| Tool / Solution | Function / Purpose | Representative Use Case |
|---|---|---|
| VASP | First-principles DFT code using PAW pseudopotentials [4]. | Calculating size-dependent cohesive energies of transition-metal nanoclusters [4]. |
| ADF | DFT software specializing in chemical bonding analysis [8]. | Performing bond energy decomposition analysis (Morokuma-type) [8]. |
| CASTEP | First-principles DFT code for solid-state materials [6]. | Studying cohesive energies and phase stability of perovskites like BaZrO₃ [6]. |
| Gaussian | Versatile quantum chemistry package [7]. | Calculating sequential bond energies in molecular clusters (e.g., H⁺·N₂) [7]. |
| LAMMPS | Classical molecular dynamics simulator [9]. | Calculating cohesive energy density in molecular systems using force fields [9]. |
| MEHnet | Multi-task equivariant graph neural network [5]. | High-throughput screening of molecular properties with CCSD(T)-level accuracy [5]. |
The accuracy of bond energy, enthalpy, and cohesive energy predictions is intrinsically linked to the chosen electronic structure method. While CCSD(T) remains the gold standard for small systems, its computational cost is prohibitive for larger molecules and materials. DFT offers a practical balance for many applications but suffers from functional-dependent accuracy. The emerging paradigm of machine-learning potentials trained on high-level quantum chemical data represents a transformative advance, promising to deliver benchmark accuracy across previously inaccessible length scales, thereby accelerating the discovery of new molecules and materials in fields ranging from drug development to energy storage.
Lattice energy, the energy associated with the formation of an ionic lattice from its gaseous ions, is a fundamental property that dictates the stability, solubility, and overall thermodynamics of ionic compounds [10] [11]. A precise understanding of this parameter is crucial for researchers and scientists, particularly in fields like drug development where the salt forms of active pharmaceutical ingredients can significantly impact stability and bioavailability. However, determining a single, definitive value for lattice energy is not straightforward, as it sits at the intersection of theoretical calculation and experimental derivation. This guide objectively explores the two primary approaches for determining lattice energy—the theoretical electrostatic model and the experimental Born-Haber cycle—and compares their underlying assumptions, protocols, and resulting values. Framed within a broader thesis on the accuracy of electronic structure methods for bond energies, this analysis highlights how the divergence between these values provides profound insight into the true nature of chemical bonding, often revealing significant covalent character in seemingly ionic compounds [12].
Fundamental Concepts: Defining Lattice Energy
The term "lattice energy" itself requires careful definition, as it is used in two contradictory ways in the literature. To avoid confusion, it is essential to qualify the term based on the direction of the process [10] [13].
- Lattice Dissociation Energy: The energy required to convert one mole of a solid ionic crystal into its separated gaseous ions. This process is always endothermic (positive value) [13]. For example, the lattice dissociation energy for NaCl is +787 kJ mol⁻¹ [10].
- Lattice Formation Energy: The energy released when one mole of a solid ionic crystal is formed from its separated gaseous ions. This process is always exothermic (negative value) [13]. For NaCl, this is -787 kJ mol⁻¹ [10].
For the remainder of this article, "lattice energy" will refer to the lattice formation energy, consistent with its use in many thermodynamic cycles. The magnitude of lattice energy is primarily governed by two factors [10] [13]:
- Ionic Charges: Higher charges on the ions lead to stronger electrostatic attraction. For instance, MgO (with Mg²⁺ and O²⁻) has a much larger lattice energy than NaCl (with Na⁺ and Cl⁻) [10].
- Ionic Radii: Smaller ions can get closer together, increasing the electrostatic attraction and thus the lattice energy. This is observed down groups in the periodic table, where lattice energies decrease as the ions become larger [10].
Methodological Approaches: A Comparative Framework
The determination of lattice energy follows two distinct philosophical and methodological pathways. The following diagram illustrates the logical relationship and key differences between these two primary approaches.
The Born-Haber Cycle: Experimental Derivation
The Born-Haber cycle is an application of Hess's Law that allows for the indirect determination of lattice energy from other, measurable thermochemical quantities [11] [14]. It is considered the source of the "experimental" or "actual" lattice energy value [12].
Detailed Experimental Protocol
The following workflow outlines the standard protocol for deriving the lattice energy of a generic ionic compound, MX, where M is a metal and X is a non-metal.
To calculate the lattice energy, the sum of the enthalpy changes for the indirect path (Steps 1-5) is set equal to the direct enthalpy of formation, leading to the following equation [14]:
LE = ΔHf - [ΔHsub(M) + IE(M) + 1/2D(X-X) + EA(X)]
- Example Calculation for NaCl [13]:
- ΔHf (NaCl) = -411 kJ mol⁻¹
- ΔHsub (Na) = +108 kJ mol⁻¹
- IE (Na) = +496 kJ mol⁻¹
- 1/2D (Cl-Cl) = +122 kJ mol⁻¹
- EA (Cl) = -349 kJ mol⁻¹
- LE = -411 - (108 + 496 + 122 - 349) = -788 kJ mol⁻¹
The accuracy of the final lattice energy is entirely dependent on the precision of the input thermochemical data [14].
Theoretical Calculations: Electrostatic Models
Theoretical lattice energy is calculated from first principles using a physics-based approach that models the ionic crystal as a collection of point charges interacting through electrostatic forces [13] [12]. The most refined of these models is the Born-Landé equation [14]:
ΔHlattice = (NA * M * z⁺ * z⁻ * e²) / (4 * π * ε₀ * r₀) * (1 - 1/n)
Where:
- N_A = Avogadro's number
- M = Madelung constant (depends on the crystal structure)
- z⁺, z⁻ = Charges of the cation and anion
- e = Elementary charge
- ε₀ = Permittivity of free space
- r₀ = Distance between ion centers
- n = Born exponent (related to the compressibility of the solid)
This method assumes a perfectly ionic compound with no covalent character, only electrostatic interactions between ions, and a perfect crystal lattice [12].
Data Comparison: Theoretical vs. Experimental Lattice Energies
The comparison between theoretical (calculated) and experimental (Born-Haber) lattice energies is not merely a check for accuracy, but a powerful diagnostic tool for understanding chemical bonding.
Table 1: Comparison of Theoretical and Experimental Lattice Energies for Selected Halides [12]
| Compound | Theoretical Lattice Energy (kJ mol⁻¹) | Experimental Lattice Energy (kJ mol⁻¹) | Difference (kJ mol⁻¹) | Implied Covalent Character |
|---|---|---|---|---|
| AgF | ~High | ~High | Small | Low |
| AgI | ~High | Lower | Large | High |
Interpreting the Data
- Small Difference: If the experimental value is very close to the theoretical value, the assumption of a highly ionic compound is valid. This is typical for compounds with cations of low charge density/polarizing power and anions that are small and non-polarizable (e.g., AgF, NaCl) [12].
- Large Difference: A significant discrepancy, where the experimental lattice energy is more negative (or less positive) than the theoretical value, indicates a failure of the purely ionic model. This signals significant covalent character in the bonding. This occurs with cations of high charge density/polarizing power that distort the electron cloud of large, easily polarizable anions (e.g., AgI) [12].
The Computational Chemistry Perspective: Benchmarking Electronic Structure Methods
The accuracy of computational chemistry methods for predicting bond energies can be benchmarked against experimental datasets like ExpBDE54, a benchmark of 54 experimental homolytic bond dissociation enthalpies (BDEs) for small molecules [15]. This is directly analogous to using Born-Haber cycles to benchmark theoretical lattice energies.
Table 2: Performance of Selected Computational Methods for BDE Prediction (ExpBDE54 Benchmark) [15]
| Computational Method | Class | Speed (Relative) | Accuracy (RMSE, kcal mol⁻¹) |
|---|---|---|---|
| g-xTB//GFN2-xTB | Semiempirical | Fastest | 4.7 |
| OMol25's eSEN | Neural Network Potential | Fast | 3.6 |
| r²SCAN-3c//GFN2-xTB | Meta-GGA DFT | Medium | ~4.0 |
| r²SCAN-D4/def2-TZVPPD | Meta-GGA DFT | Slow | 3.6 |
| ωB97M-D3BJ/def2-TZVPPD | Hybrid DFT | Slow | 3.7 |
Key Insights from Computational Benchmarking
- Linear Regression Corrections: Raw electronic bond dissociation energies (eBDEs) from computational methods show a strong linear relationship with experimental BDEs but require empirical correction for zero-point energy, enthalpy, and relativistic effects to achieve high accuracy [15].
- Pareto Frontier of Methods: The benchmarking identifies a "Pareto frontier" of methods that offer the best trade-off between speed and accuracy. For rapid screening on CPU, semiempirical methods like g-xTB//GFN2-xTB are optimal, while for higher accuracy, neural network potentials like eSEN or composite DFT methods like r²SCAN-3c represent the best choices [15].
- Basis Set Convergence: For density functional theory (DFT) methods, moving to larger basis sets beyond def2-TZVPPD offers negligible improvement in BDE accuracy while significantly increasing computational cost, suggesting the limit of a purely electronic energy approach has been nearly reached [15].
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key solutions and materials essential for research in experimental thermochemistry and computational modeling of bond energies.
Table 3: Essential Research Reagent Solutions and Materials
| Item | Function & Application |
|---|---|
| High-Purity Metal & Gas Samples | Essential for accurate calorimetric measurements of standard enthalpies of formation (ΔH_f). Impurities lead to significant errors in Born-Haber cycles. |
| Calorimetry Apparatus | The primary experimental setup for directly measuring heat changes (e.g., enthalpy of formation, sublimation, and solution) required for Born-Haber cycles. |
| Mass Spectrometer | Used in conjunction with Knudsen effusion cells for vapor pressure measurements, crucial for determining accurate sublimation enthalpies. |
| Quantum Chemistry Software | Platforms like PSI4 and Gaussian are used for computational determination of bond energies and theoretical lattice energies via electronic structure methods [15]. |
| Semiempirical & Neural Network Codes | Software such as xtb (for GFNn-xTB methods) and implementations for neural network potentials (eSEN, UMA) enable high-throughput screening of bond strengths [15]. |
| Benchmark Datasets (e.g., ExpBDE54) | Curated sets of reliable experimental data serve as the essential ground truth for validating and refining the accuracy of computational methods [15]. |
Applications and Implications in Materials Science and Drug Development
The principles of lattice energy and bond strength quantification extend far beyond simple salts, providing a foundation for understanding and designing complex materials.
- Predicting Ionic Compound Stability and Properties: Lattice energy directly correlates with key physical properties. A high lattice energy generally results in high melting and boiling points, low solubility in polar solvents, and increased hardness and brittleness [14]. This understanding is critical for selecting appropriate salt forms in pharmaceutical development to optimize stability and bioavailability.
- From Atomic Bonding to Macroscopic Strength in Metals: Recent research demonstrates a direct correlation between bond strength, quantified by cohesive energy (Ecoh), and the macroscopic mechanical properties of metals and multi-principal-element alloys (MPEAs) [16]. Models have been developed that use Ecoh and atomic radius to predict grain-boundary energies, which in turn control the material's strength according to the Hall-Petch relationship [16]. This provides a physical picture linking atomic-scale bond strength to macro-scale properties for materials design.
The interplay between theoretical and experimental lattice energy, mediated by the Born-Haber cycle, remains a cornerstone of quantitative chemistry. The divergence between these values is not a failure of theory, but a successful diagnostic that reveals the nuanced reality of chemical bonding, where pure ionic character is often an idealization. For researchers and drug development professionals, this comparative framework is indispensable. It provides a rigorous method for validating computational models against experimental benchmarks, ensures the accurate prediction of material properties, and guides the rational selection of compounds with desired stability and performance characteristics. As computational methods advance, the synergy between high-accuracy quantum chemistry and reliable experimental thermochemical cycles will continue to deepen our understanding of bond energies and accelerate the design of novel materials and pharmaceuticals.
The chemisorption energy, representing the bond strength between an adsorbate and a material surface, serves as a fundamental determinant in numerous chemical processes ranging from heterogeneous catalysis to corrosion and nanotechnology [17]. Accurate prediction of this property enables researchers to design surfaces with optimal characteristics, thereby accelerating the development of efficient catalysts, durable materials, and novel functional interfaces. Electronic-structure-based models have emerged as indispensable tools for this purpose, providing a physical framework to interpret and predict adsorption strengths without resorting to exhaustive experimental testing. Among these, the d-band model pioneered by Hammer and Nørskov has achieved notable success, establishing the d-band center (the average energy of d-states relative to the Fermi level) as a central descriptor for trends in chemisorption strength across transition metal surfaces [17]. This model effectively correlates electronic structure features obtained before interaction with the resulting chemisorption energy, offering a powerful simplifying principle for surface science.
However, the increasing complexity of modern materials—including multi-metallic alloys, intermetallics, and high-entropy alloys—has revealed limitations in conventional d-band center approaches [17]. These shortcomings primarily arise because the d-band center alone carries no information about band dispersion, asymmetries, or distortions in the electronic structure introduced by alloying, and fails to fully account for perturbations in the surface electronic states induced by the adsorbate itself. Consequently, researchers have developed enhanced models that incorporate additional electronic factors beyond the d-band center, such as d-band width, higher moments of the d-band, and coordination effects, to achieve improved accuracy across broader ranges of material systems. This guide provides a comprehensive comparison of these electronic structure factors, evaluating their predictive performance, methodological requirements, and applicability for contemporary challenges in surface chemistry and catalysis research.
Theoretical Framework: From d-Band Center to Advanced Descriptors
Foundations of the d-Band Model
The conventional d-band model operates on the principle that chemisorption energy (ΔE) can be decomposed into contributions from interaction with the metal sp-electrons and the d-electrons: ΔE = ΔEsp + ΔEd [17]. The sp-electron contribution is typically large and attractive but approximately constant across transition metals, while the d-electron contribution varies systematically and primarily governs trends in adsorption strength. In this framework, the d-band center (ε_d) serves as the principal descriptor, where a higher-lying d-band center (closer to the Fermi level) generally correlates with stronger bonding. This occurs because a higher d-band center enhances coupling with adsorbate states and shifts the antibonding states to higher energies, potentially above the Fermi level, resulting in increased occupancy of bonding states and stronger adsorption [17]. The model has demonstrated remarkable success in explaining trends across pure transition metals and some simple alloys, establishing itself as a foundational concept in surface chemistry.
The physical basis for this model originates from the Newns-Anderson approach, which describes the interaction between a single adsorbate energy level and the continuum of surface electronic states [17]. In transition metals, the localized d-states with their narrow energy distribution interact with the adsorbate level to produce bonding and antibonding states, while the broad, delocalized sp-states produce a single renormalized resonance. The simplicity and intuitive nature of the d-band model have contributed to its widespread adoption, though its limitations in treating complex alloys have motivated the development of more sophisticated approaches that capture additional electronic structure features beyond the d-band center alone.
Advanced Electronic Structure Descriptors
Recent research has identified several electronic structure factors that enhance predictive capability beyond the basic d-band center approach. These advanced descriptors address specific limitations of the conventional model, particularly for multi-metallic systems:
d-Band Width and Higher Moments: The d-band width, derived from the second moment of the d-band density of states, provides information about the dispersion and coordination environment of surface atoms [18]. Incorporating this descriptor helps account for variations in local coordination geometry that significantly impact chemisorption behavior. Some advanced models also utilize the d-band skewness (third moment) and kurtosis (fourth moment) to capture asymmetries and peak shapes in the d-band density of states that influence bonding interactions [17].
d-Band Filling: The occupation of d-states plays a critical role in determining chemisorption strength, as it affects the electron transfer capabilities and the position of antibonding states relative to the Fermi level [17] [19]. Systems with high d-band filling typically exhibit weaker adsorption due to increased occupation of antibonding states, while lower d-band filling often correlates with stronger bonding.
Adsorbate-Induced Effects: Advanced models recognize that adsorbates not only perturb surface electronic states but also induce changes in the adsorption site that interact with the chemical environment [17]. This leads to a second-order response in chemisorption energy with the d-filling of neighboring atoms, explaining deviations from simple linear behavior observed in complex alloys.
Non-ab Initio Descriptors: For large-scale screening, researchers have developed descriptors that do not require density functional theory calculations, such as combining d-band width from muffin-tin orbital theory with electronegativity to account for adsorbate renormalization [18]. These enable rapid first-pass screening of materials while maintaining reasonable accuracy.
Table 1: Key Electronic Structure Descriptors in Chemisorption Models
| Descriptor | Physical Significance | Strengths | Limitations |
|---|---|---|---|
| d-Band Center | Average energy of d-states relative to Fermi level | Intuitive; good for trend prediction across pure metals | Neglects band shape and width; inadequate for complex alloys |
| d-Band Width | Measure of d-band dispersion related to coordination | Accounts for local coordination environment | Requires additional calculation; interpretation less straightforward |
| d-Band Filling | Occupation of d-states | Captives electron transfer capabilities; affects antibonding occupancy | Interplays with other factors in complex ways |
| Higher Moments | Shape and asymmetry of d-band distribution | Captures nuanced features of electronic structure | Computationally intensive; complex interpretation |
| Electronegativity | Tendency to attract electrons in chemical bonds | Simple descriptor for adsorbate effects | Oversimplifies complex charge transfer processes |
Performance Comparison of Electronic Structure Methods
Accuracy Assessment Across Methodologies
The predictive accuracy of electronic structure methods varies significantly based on the complexity of the descriptors employed and the material systems under investigation. Quantitative comparisons reveal distinct performance patterns across different approaches:
Traditional d-band center models typically achieve mean absolute errors (MAEs) of approximately 0.15-0.25 eV for adsorption energies on pure transition metals, but these errors increase substantially for bimetallic and multi-component systems, sometimes exceeding 0.5 eV [17]. This degradation in performance highlights the fundamental limitations of relying solely on the d-band center for complex alloys. In contrast, advanced models incorporating multiple electronic factors demonstrate markedly improved accuracy. For instance, models employing d-band width plus electronegativity as descriptors have achieved MAEs of 0.05 eV for CO adsorption on 263 alloy systems when combined with active learning algorithms [18]. Without active learning, the accuracy decreased to 0.18 eV, underscoring the importance of sampling strategy in addition to descriptor selection.
Recent physics-based models employing first and second moments of the d-band along with d-band filling have demonstrated robust performance across diverse systems, reporting MAEs of 0.13 eV versus density functional theory reference values for O, N, CH, and Li chemisorption on bi- and tri-metallic surface and subsurface alloys [17]. This represents a significant improvement over conventional d-band center approaches while maintaining physical interpretability. The integration of machine learning methods with electronic structure descriptors has further enhanced predictive capability, with neural network (NN) and kernel ridge regression (KRR) approaches successfully mapping descriptors to adsorption energies while preserving computational efficiency compared to direct quantum calculations [18].
Application-Specific Performance Considerations
The optimal choice of electronic structure model depends critically on the specific application requirements, including material complexity, desired accuracy, and computational constraints:
For high-throughput screening of bimetallic catalysts, models combining d-band features with coordination numbers or electronegativity often provide the best balance between computational cost and predictive accuracy [18] [19]. These approaches have successfully identified promising catalyst formulations, such as Cu₃Y@Cu for electrochemical CO₂ reduction with an overpotential approximately 1 V lower than gold catalysts [18]. For fundamental studies of adsorption mechanisms on well-defined surfaces, more sophisticated approaches incorporating higher moments of the d-band may be justified despite their increased computational demands, as they provide deeper physical insight into bonding interactions [17].
In applications requiring extreme accuracy for small systems, high-level quantum chemistry methods like CCSD(T) remain the gold standard, though they are computationally prohibitive for most practical catalyst screening applications [20] [21]. For example, CCSD(T) calculations have demonstrated remarkable accuracy for bond dissociation energies in small molecules, with errors potentially below 1 kcal/mol (0.043 eV) when using appropriate basis sets and accounting for core-valence correlation effects [20]. However, such methods remain impractical for surface systems of meaningful size, necessitating the continued development and use of descriptor-based models.
Table 2: Performance Comparison of Electronic Structure Methods for Bond Energy Prediction
| Method Category | Representative Methods | Typical MAE Range | Computational Cost | Ideal Use Cases |
|---|---|---|---|---|
| Classic d-Band Center | Hammer-Nørskov model | 0.15-0.25 eV (higher for alloys) | Low | Trend analysis on pure metals; educational contexts |
| Multi-Descriptor d-Band | d-band center + width + filling | 0.05-0.15 eV | Moderate | Alloy catalyst screening; surface design |
| Machine Learning Enhanced | NN/KRR with electronic descriptors | 0.05-0.10 eV (with active learning) | Low (after training) | High-throughput screening of complex materials |
| High-Level Quantum Chemistry | CCSD(T), (RO)CBS-QB3 | <0.05 eV for small molecules | Very High | Benchmark calculations; method validation |
| Density Functional Theory | Various functionals (e.g., B3LYP, PBE, r²SCAN) | 0.03-0.20 eV (functional-dependent) | Moderate to High | Direct adsorption energy calculation; training data generation |
Experimental Protocols and Methodologies
Computational Workflows for Descriptor Evaluation
The reliable calculation of electronic structure descriptors requires carefully designed computational protocols. For surface slab models, standard approaches typically employ density functional theory with generalized gradient approximation (GGA) functionals such as PBE, which provide reasonable accuracy for metallic systems at manageable computational cost [19]. The d-band center is calculated as the first moment of the projected d-band density of states: εd = ∫{-∞}^{EF} ε nd(ε)dε / ∫{-∞}^{EF} nd(ε)dε, where nd(ε) represents the density of d-states at energy ε, and E_F is the Fermi energy [19]. For magnetic systems, separate d-band centers must be computed for spin-up and spin-down channels, as significant differences can dramatically influence adsorption behavior [19].
Higher moments of the d-band follow analogous definitions: the width (second moment) is calculated as Wd = [∫{-∞}^{EF} (ε-εd)² nd(ε)dε / ∫{-∞}^{EF} nd(ε)dε]^{1/2}, while the skewness (third moment) and kurtosis (fourth moment) provide information about distribution asymmetry and peak sharpness, respectively [17]. These calculations typically employ specialized codes such as VASP with post-processing tools like VASPKIT for electronic structure analysis [19]. For high-quality results, computational parameters must be carefully controlled, including using appropriate k-point meshes for Brillouin zone sampling (e.g., 8×8×1 for surface calculations), sufficient plane-wave cutoff energy (typically 400-520 eV), and proper treatment of core electrons using the projector augmented-wave (PAW) method [19].
Figure 1: Computational workflow for descriptor-based chemisorption prediction
Benchmarking and Validation Approaches
Rigorous validation of chemisorption models requires comparison against high-quality reference data, typically obtained from carefully converged DFT calculations or experimental measurements where available. Standard benchmarking protocols involve calculating adsorption energies for a diverse set of adsorbate-surface combinations and comparing predictions against reference values using statistical metrics such as mean absolute error (MAE), root mean square error (RMSE), and correlation coefficients [17] [21].
For method development, established benchmark datasets like BSE49 (comprising 4,502 bond-separation energy values computed at the (RO)CBS-QB3 level) provide valuable references for assessing performance across diverse chemical systems [21]. Similarly, experimental benchmark sets such as ExpBDE54 offer gas-phase bond dissociation enthalpies for validation, though appropriate corrections must be applied to account for zero-point energy, enthalpy, and relativistic effects when comparing with computational results [15]. Active learning approaches significantly enhance validation efficiency by strategically selecting training points that maximize information gain, thereby reducing the number of expensive reference calculations required to achieve target accuracy levels [18].
When validating models for surface adsorption, it is essential to consider various potential error sources, including basis set superposition error (BSSE), which can be corrected using the counterpoise method of Boys and Bernardi [22]. For systems with potential multireference character, such as those involving bond dissociation, methods beyond single-reference DFT may be necessary, including complete active space SCF (CASSCF) or multi-reference configuration interaction (MRCI) approaches [22]. These considerations ensure robust validation and prevent overestimation of model performance due to systematic computational errors.
Essential Research Reagents and Computational Tools
Software Solutions for Electronic Structure Analysis
The computational study of electronic structure factors in chemisorption relies on specialized software tools spanning from ab initio calculation to descriptor analysis and machine learning:
VASP (Vienna Ab initio Simulation Package): A widely used package for DFT calculations employing the projector augmented-wave (PAW) method [19]. Essential for calculating surface electronic structures, optimizing adsorption geometries, and computing d-band properties using GGA functionals like PBE.
Gaussian: A comprehensive quantum chemistry package supporting various methods from Hartree-Fock to coupled cluster theory and density functional theory [21]. Particularly valuable for benchmarking studies on cluster models and calculating accurate reference data using composite methods like CBS-QB3.
MOLPRO: A specialized quantum chemistry software with strengths in high-accuracy multireference methods, including CASSCF, MRCI, and coupled cluster techniques [22]. Useful for studying systems with strong electron correlation or multireference character where standard DFT fails.
VASPKIT: A post-processing toolkit for VASP that automates the calculation of electronic structure descriptors, including d-band centers, widths, and higher moments [19]. Streamlines the extraction of chemisorption-relevant properties from DFT calculations.
xtb: A semiempirical quantum chemistry program providing fast calculations using the GFNn-xTB methods [15]. Enables rapid geometry optimizations and preliminary screening studies at a fraction of the cost of DFT.
Table 3: Essential Computational Tools for Chemisorption Studies
| Tool | Primary Function | Key Features | Typical Use Case |
|---|---|---|---|
| VASP | DFT Calculations | PAW method; surface modeling; DOS analysis | First-principles surface and adsorption studies |
| Gaussian | Quantum Chemistry | Composite methods; molecular calculations | Benchmarking; cluster model studies |
| psi4 | Quantum Chemistry | Efficient DFT implementation; various wavefunction methods | Method development; testing new functionals |
| VASPKIT | Post-processing | Automated descriptor calculation; data extraction | Streamlining analysis of VASP results |
| xtb | Semiempirical Methods | Fast geometry optimization; large systems | Preliminary screening; conformational analysis |
High-quality reference data is essential for developing and validating new chemisorption models. Several curated datasets serve as valuable resources for the research community:
BSE49 Dataset: A comprehensive collection of 4,502 bond-separation energies for 49 unique bond types calculated at the (RO)CBS-QB3 level of theory [21]. Provides non-relativistic ground-state energy differences without zero-point corrections, ideal for testing electronic structure methods.
ExpBDE54: A curated set of experimental homolytic bond-dissociation enthalpies for 54 small molecules, focusing on carbon-hydrogen and carbon-halogen bonds most relevant to organic and medicinal chemistry [15]. Useful for validating methods against experimental measurements.
Catalyst Datasets: Specialized collections of adsorption energies on various metal and alloy surfaces, typically computed using DFT [18] [17]. Enable direct testing of chemisorption models for catalytic applications.
These resources facilitate standardized comparisons between different electronic structure methods and ensure that performance claims are based on consistent, reproducible benchmarks. When using these datasets, researchers should adhere to the intended applications—for instance, BSE49 is designed for testing electronic energy calculations rather than direct comparison with experimental bond dissociation enthalpies, which require additional corrections for zero-point energy and thermal effects [21].
Electronic structure factors governing chemisorption have evolved significantly beyond the conventional d-band center model, with advanced descriptors incorporating d-band width, filling, and higher moments demonstrating markedly improved accuracy for complex material systems. The comparative analysis presented in this guide reveals a clear trade-off between model complexity and predictive performance, with multi-descriptor approaches achieving mean absolute errors as low as 0.05-0.13 eV for diverse adsorbates on alloy surfaces [18] [17]. These developments address critical limitations of traditional models while maintaining physical interpretability—an essential consideration for guiding materials design.
Future advancements in this field will likely emerge from several promising directions. The integration of machine learning with electronic structure descriptors represents a particularly powerful approach, combining physical insight with data-driven pattern recognition to navigate complex materials spaces [18] [21]. Additionally, increasing attention to adsorbate-induced surface perturbations and their interaction with the local chemical environment will further refine our understanding of bonding interactions in complex systems [17]. The development of efficient non-ab initio descriptors will continue to enable high-throughput screening, while benchmark datasets and active learning strategies will optimize the use of computational resources for model training and validation [18] [21] [15].
For researchers and drug development professionals, these methodological advances translate to increasingly reliable tools for predicting surface interactions and designing optimized materials. As electronic structure methods continue to evolve, their capacity to guide experimental efforts will further strengthen, accelerating the discovery of next-generation catalysts, functional materials, and therapeutic agents through computationally driven design.
For researchers in drug development and materials science, accurately predicting the strength of chemical bonds is not an academic exercise—it is a fundamental prerequisite for understanding stability, reactivity, and metabolic fate. Experimental gas-phase measurements provide the indispensable foundation for this understanding, serving as the highest standard for benchmarking the computational methods used in molecular design [23]. By isolating molecules from the complicating effects of solvents and counter-ions, these measurements yield precise dissociation energies that reflect the true, unperturbed intermolecular interaction strengths [23].
This guide objectively compares key experimental benchmarks and the electronic structure methods they validate. It provides researchers with the data needed to select the most appropriate computational workflow for predicting bond energies, a critical task in applications ranging from predicting sites of drug metabolism to designing new catalysts.
Key Experimental Benchmark Datasets
Several high-quality datasets serve as standardized references for validating computational chemistry methods. The table below summarizes two prominent benchmarks for bond energy data.
Table 1: Comparison of Key Experimental and Computational Benchmark Datasets
| Dataset Name | ExpBDE54 [15] | BSE49 [21] |
|---|---|---|
| Data Origin | Experimental gas-phase measurements | High-level theoretical computations ((RO)CBS-QB3) |
| Data Content | 54 experimental homolytic Bond-Dissociation Enthalpies (BDEs) | 4,502 Bond Separation Energies (BSEs) for 49 unique bond types |
| Primary Utility | External validation of computational methods | Training and parametrization of lower-cost methods |
| Key Features | Covers C-H and C-halogen bonds relevant to organic & medicinal chemistry | Extensive and diverse, includes "Existing" and "Hypothetical" molecules |
| Direct Experimental Comparison | Yes, provides benchmark BDE values | No, values are not directly comparable to experimental BDEs |
The ExpBDE54 dataset is a "slim" benchmark curated from experimental gas-phase studies, ideal for quickly testing a method's performance on chemically relevant bonds [15]. In contrast, the BSE49 dataset is a large, computationally derived resource ideal for training machine learning potentials or parameterizing semi-empirical methods, though its energies lack zero-point and thermal corrections [21].
Benchmarking Electronic Structure Methods
The true value of experimental benchmarks lies in their ability to evaluate the accuracy and efficiency of computational workflows. A 2025 study leveraged the ExpBDE54 dataset to map the Pareto frontier of bond-dissociation-enthalpy-prediction methods, balancing speed against accuracy [15].
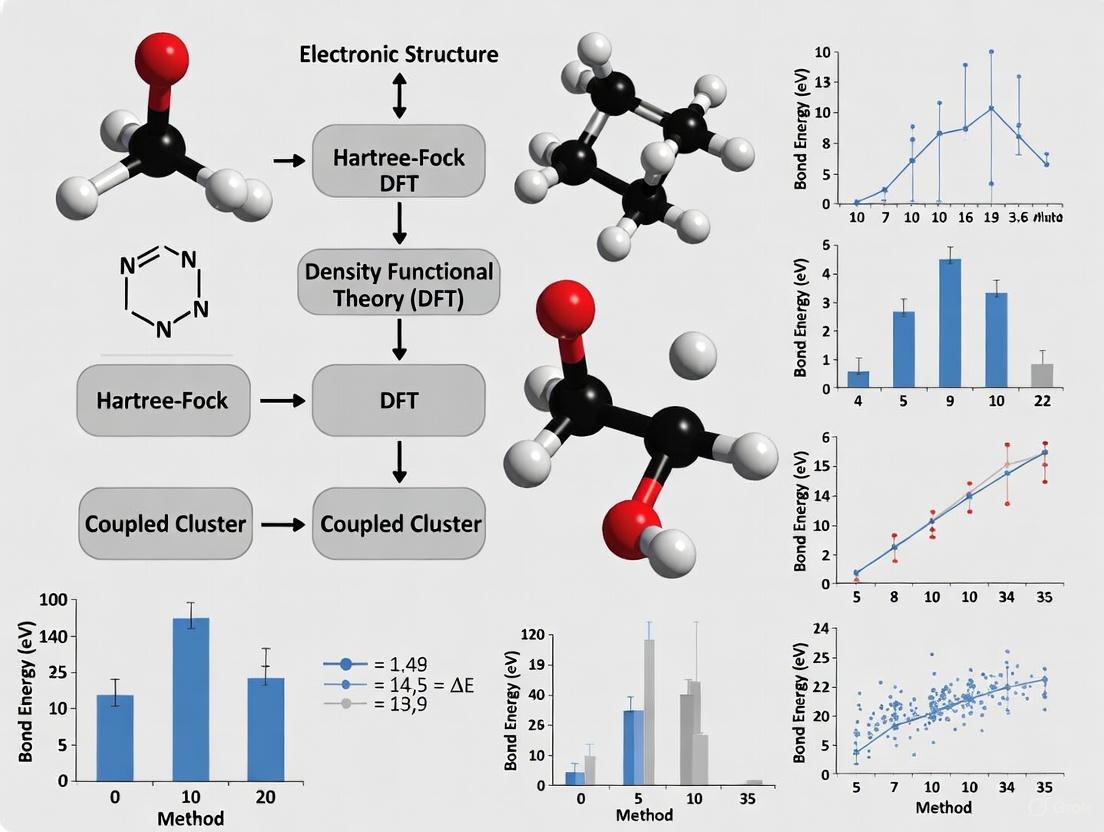
Table 2: Performance of Selected Computational Methods on the ExpBDE54 Benchmark (Root-Mean-Square Error in kcal·mol⁻¹) [15]
| Methodology Class | Specific Method | Reported RMSE | Key Application Context |
|---|---|---|---|
| Neural Network Potential | OMol25's eSEN Conserving Small | 3.6 | Medium-sized systems where accuracy is prioritized [15] |
| Semiempirical (GFN2-xTB optimized) | g-xTB//GFN2-xTB | 4.7 | CPU-based calculations where speed is critical [15] |
| Density-Functional Theory (DFT) | r2SCAN-D4/def2-TZVPPD | 3.6 | High-accuracy reference standard [15] |
| DFT with Semiempirical Optimization | r2SCAN-3c//GFN2-xTB | Best speed/accuracy tradeoff for a QM-based method [15] |
The benchmarking reveals that suitably corrected semiempirical and machine-learning approaches can rival the accuracy of more expensive DFT methods at a fraction of the computational cost. For instance, g-xTB//GFN2-xTB offers a rapid solution on CPU hardware, while OMol25's eSEN Conserving Small neural network potential achieves top-tier accuracy for medium-sized systems [15]. The composite DFT method r2SCAN-3c provides an excellent balance of speed and accuracy, especially when paired with a GFN2-xTB geometry optimization [15].
Detailed Experimental Protocols
The SEP-R2PI Method for Intermolecular Dissociation Energies
A premier experimental technique for obtaining gas-phase ground-state dissociation energies (D₀(S₀)) of cold, isolated bimolecular complexes is the SEP-R2PI (Stimulated Emission Pumping-Resonant Two-Photon Ionization) method [23]. This laser-based approach allows for the precise measurement of the dissociation energy for noncovalent complexes (e.g., M⋅S, where M is an aromatic molecule and S is a solvent atom/molecule) with bracketing accuracies of ≤1.0 kJ/mol [23].
The workflow involves a triply resonant process that deposits energy directly into the ground-state vibrational modes of the complex to induce dissociation.
Diagram 1: SEP-R2PI Method Workflow. This laser technique precisely measures gas-phase dissociation energies.
This protocol has been successfully applied to characterize 55 different M⋅S complexes, creating a large experimental database for understanding noncovalent interactions like hydrogen bonding and London dispersion [23].
Workflow for Computational Benchmarking
To validate a computational method against a benchmark like ExpBDE54, a standardized workflow is followed to calculate electronic bond-dissociation energies (eBDEs) and compare them to experimental enthalpies.
Diagram 2: Computational BDE Benchmarking Workflow. This process calculates theoretical bond dissociation energies for comparison with experimental benchmarks.
The linear regression correction in the final step is crucial, as it accounts for systematic errors and missing contributions like zero-point energy and enthalpic effects, enabling a fair comparison between computed electronic energies and experimental enthalpies [15].
The Scientist's Toolkit
The following table details key resources and methodologies essential for work in this field.
Table 3: Essential Research Reagents and Computational Tools
| Tool Name | Type | Primary Function in Research |
|---|---|---|
| SEP-R2PI Laser Spectroscopy [23] | Experimental Apparatus | Provides gold-standard gas-phase intermolecular dissociation energies (D₀) for benchmark complexes. |
| ExpBDE54 Dataset [15] [24] | Experimental Benchmark Data | Serves as a "slim" external benchmark for validating BDE-prediction methods on 54 small molecules. |
| BSE49 Dataset [21] | Computational Reference Data | Provides 4,502 high-quality theoretical Bond Separation Energies for method training and development. |
| GFN2-xTB [15] | Semiempirical Quantum Method | Used for fast geometry optimizations of parent molecules and radical fragments in computational workflows. |
| r2SCAN-3c [15] | Density Functional Theory (DFT) | A "Swiss-army knife" DFT method offering a strong balance of accuracy and computational speed for BDE prediction. |
| g-xTB [15] | Semiempirical Quantum Method | A low-cost method suitable for rapid BDE calculations, especially when on CPU and speed is a premium. |
Gas-phase experimental measurements provide the non-negotiable standard for accuracy in bond energy research. Datasets like ExpBDE54 offer a critical foundation for objectively comparing and improving computational methods, from fast semiempirical approaches to high-level DFT and neural network potentials [15]. For researchers in drug development, leveraging these benchmarks enables more reliable predictions of metabolic stability and reactivity, ultimately informing the design of safer and more effective therapeutics. As computational power grows and methods evolve, the role of precise, gas-phase experimental data will only become more vital in pushing the frontiers of accuracy in electronic structure theory.
Computational Toolkit: A Practical Guide to DFT, Semiempirical, and Machine Learning Methods
Within computational chemistry, predicting bond dissociation energies (BDEs) with high accuracy is fundamental to understanding reaction mechanisms, catalyst design, and predicting metabolic sites in drug development. While highly accurate wavefunction methods exist, their computational cost often precludes their use for large, chemically relevant systems. Consequently, Kohn-Sham Density Functional Theory (KS-DFT) remains the workhorse for such applications, with its accuracy critically dependent on the chosen exchange-correlation functional. This guide objectively compares the performance of three popular density functional approximations—r2SCAN-D4, ωB97M-D3BJ, and B3LYP-D4—in the context of bond energy calculations, providing researchers with the experimental data and protocols needed to make an informed selection.
Theoretical Background and Functional Profiles
The selected functionals represent different rungs on Perdew's "Jacob's Ladder" of DFT approximations, each with a distinct approach to incorporating physical constraints and empirical data.
r2SCAN-D4: A meta-Generalized Gradient Approximation (meta-GGA) that is "regularized" to restore the formal constraints of its predecessor, SCAN, while achieving superior numerical stability. It is typically combined with the modern D4 dispersion correction [25] [26]. Its key strength is its non-empirical design, offering high transferability without fitting parameters, and it approaches the accuracy of hybrid functionals while retaining the speed of a semi-local functional [25].
ωB97M-D3BJ: A range-separated hybrid meta-GGA, meaning it uses exact Hartree-Fock exchange at long interelectronic ranges and a meta-GGA at short ranges. It is routinely paired with the D3(BJ) empirical dispersion correction. This functional is known for its high performance across a wide range of properties, including thermochemistry and non-covalent interactions [27].
B3LYP-D4: A global hybrid GGA, one of the most widely used functionals in computational chemistry. It mixes a fixed percentage of exact exchange with GGA exchange and correlation. The addition of the latest D4 dispersion correction modernizes this classic functional, improving its description of non-covalent interactions [28].
Performance Benchmarking on Bond Dissociation Energies
The accuracy of these functionals for bond energy predictions is quantitatively assessed using the ExpBDE54 dataset, a compilation of 54 experimental homolytic bond-dissociation enthalpies for carbon-hydrogen and carbon-halogen bonds [15].
Table 1: Performance of DFT Functionals on the ExpBDE54 Benchmark Set (RMSE in kcal/mol)
| Functional | Basis Set | RMSE (ExpBDE54) | Computational Cost |
|---|---|---|---|
| r2SCAN-D4 | def2-TZVPPD | 3.6 | Benchmark |
| ωB97M-D3BJ | def2-TZVPPD | 3.7 | Comparable to r2SCAN-D4 |
| B3LYP-D4 | def2-TZVPPD | 4.1 | ~2x Faster than r2SCAN-D4 |
| r2SCAN-D4 | vDZP | ~5.1 | ~2x Faster than def2-TZVPPD |
| ωB97M-D3BJ | vDZP | ~4.8 (Most Accurate with vDZP) | ~2x Faster than def2-TZVPPD |
The data reveals several key trends [15]:
- r2SCAN-D4/def2-TZVPPD is the most accurate combination overall, achieving an RMSE of 3.6 kcal/mol.
- Moving to the more efficient vDZP basis set, ωB97M-D3BJ becomes the most accurate functional, demonstrating its robustness with smaller basis sets.
- B3LYP-D4 offers a attractive balance, providing respectable accuracy (4.1 kcal/mol) with a significant speed advantage, calculated to be approximately twice as fast as the r2SCAN-D4/def2-TZVPPD workflow.
Performance Across Diverse Chemical Properties
Beyond BDEs, the overall utility of a functional depends on its performance across various chemical properties. The extensive GMTKN55 database, which encompasses main-group thermochemistry, kinetics, and non-covalent interactions, provides a broad view of functional reliability.
Table 2: Generalized Performance on the GMTKN55 Database (WTMAD2 in kcal/mol)
| Functional | Overall WTMAD2 (GMTKN55) | Barrier Heights | Non-Covalent Interactions | Transition Metal Chemistry |
|---|---|---|---|---|
| r2SCAN-D4 | 7.5 [25] | Good | Good | Excellent [29] [30] |
| ωB97M-D3BJ | Not Explicitly Reported | Excellent [27] | Excellent [31] [27] | Good |
| B3LYP-D4 | 6.4 (with vDZP) [28] | Moderate | Moderate | Moderate [29] |
Key insights from this broader benchmarking include [29] [30] [31]:
- r2SCAN-D4 shows exceptional versatility, particularly in transition metal chemistry. It is a top performer for the geometry of spin-coupled iron-sulfur clusters like the nitrogenase FeMoco cluster, with errors for metal-metal distances of just 0.8% [30] [25].
- ωB97M-D3BJ excels in kinetics and non-covalent interactions. It is a top-tier range-separated hybrid for calculating barrier heights and reaction energies and performs exceptionally well for hydrogen-bonding energies in supramolecular complexes [31] [27].
- B3LYP-D4, while less specialized, delivers solid all-around performance, especially when paired with an efficient basis set like vDZP [28].
Detailed Computational Protocols
To ensure reproducibility, this section outlines the standard computational protocols used in the benchmark studies cited herein.
Bond Dissociation Enthalpy (BDE) Calculation Workflow
The following diagram illustrates the end-to-end workflow for computing accurate BDEs, adapted from benchmark studies [15].
Key Steps in the BDE Calculation Protocol [15]:
- Initial Geometry Optimization: Molecular geometries are generated from SMILES strings and optimized. For efficiency, the semi-empirical GFN2-xTB method is often used, though DFT optimization can also be employed.
- Single-Point Energy on Intact Molecule: A high-level DFT single-point energy calculation is performed on the optimized geometry.
- Bond Cleavage and Fragment Optimization: The target bond is broken homolytically, generating two doublet radical fragments. These fragments are then geometry-optimized.
- Single-Point Energy on Fragments: High-level DFT single-point energy calculations are performed on the optimized fragments.
- Electronic BDE (eBDE) Calculation: The eBDE is computed as the energy difference between the fragments and the parent molecule.
- Empirical Correction: A linear regression correction is applied to the eBDEs to account for the lack of zero-point energy, enthalpic, and relativistic effects, bringing the values into agreement with experimental BDEs.
General DFT Single-Point Energy Protocol
The following settings represent a robust standard for obtaining accurate DFT energies, as used in modern benchmarks [28] [15] [31].
- Software: Calculations are typically performed with quantum chemistry packages like Psi4 or ORCA.
- Integration Grid: A dense integration grid is crucial for meta-GGA and hybrid functionals. Common choices include a (99,590) grid or the
DEFGRID3setting in ORCA [28] [26]. - Dispersion Correction: Empirical dispersion corrections (D3(BJ) or D4) must be included, as they are essential for non-covalent interactions and accurate thermochemistry [31] [25].
- Basis Set Selection:
- SCF Convergence: Techniques like a level shift (e.g., 0.10 Hartree) and tight integral tolerances (e.g., 10⁻¹⁴) are used to ensure robust self-consistent field convergence [28].
Table 3: Key Software, Basis Sets, and Benchmarks for DFT Studies
| Tool Name | Category | Primary Function & Application |
|---|---|---|
| Psi4/ORCA | Software | Open-source quantum chemistry packages for running DFT, HF, and correlated calculations. |
| def2-TZVPPD | Basis Set | Triple-zeta quality basis with diffuse functions; used for highly accurate single-point energies. |
| vDZP | Basis Set | Pareto-efficient double-zeta basis for rapid calculations with low basis-set error [28]. |
| GMTKN55 | Benchmark | Database of 55 main-group chemical problems for general functional benchmarking [25] [26]. |
| ExpBDE54 | Benchmark | A "slim" benchmark of 54 experimental BDEs for validating bond-strength prediction methods [15]. |
| GFN2-xTB | Software | Semi-empirical method for fast geometry optimizations, often used as a precursor to DFT single-points [15]. |
| D4/D3(BJ) | Correction | Empirical dispersion corrections that must be added to functionals for physical interaction energies. |
The experimental data clearly indicates that there is no single "best" functional for all scenarios; the optimal choice depends on the specific chemical problem, system size, and required accuracy.
- For Maximum BDE Accuracy: The r2SCAN-D4/def2-TZVPPD combination is the top performer, making it the ideal choice for definitive studies on bond strengths where computational cost is secondary to accuracy.
- For a Balance of Speed and Accuracy: ωB97M-D3BJ/vDZP provides an excellent compromise, being the most accurate functional with the efficient vDZP basis set for BDEs. It is also the preferred choice for reactions involving significant non-covalent interactions or complex barrier heights.
- For Large Systems and Routine Screening: B3LYP-D4/vDZP offers a reliable and computationally efficient option for screening large libraries of compounds, such as in drug discovery for predicting metabolic sites, while still providing good quantitative accuracy.
For systems containing transition metals or complex spin-coupled clusters like iron-sulfur cofactors, r2SCAN-D4 and its hybrid variants are particularly recommended due to their superior description of metal-ligand covalency and geometric structure [29] [30]. Researchers are encouraged to use the provided protocols and benchmarks to validate these functionals for their specific applications.
In the field of computational chemistry, the demand for efficient electronic structure methods that can balance speed with accuracy is a central research focus, particularly for applications in high-throughput screening and bond energies research. Semi-empirical quantum chemistry methods, which are based on the Hartree-Fock formalism but incorporate approximations and empirical parameters, have emerged as crucial tools for treating large molecules where more accurate ab initio methods become prohibitively expensive [32]. These methods obtain some parameters from empirical data, which allows for a partial inclusion of electron correlation effects [32].
The GFN-xTB (Geometry, Frequency, Noncovalent interactions, eXtended Tight Binding) family of methods, developed by Grimme's group, represents a significant advancement in this domain. These methods are designed to provide a compelling balance between computational efficiency and accuracy across a broad spectrum of molecular properties [33]. This guide provides an objective comparison between two key members of this family—the established GFN2-xTB and the newer general-purpose g-xTB—evaluating their performance specifically for high-throughput screening applications within the context of bond energy research.
The GFN-xTB Framework
GFN-xTB methods are semi-empirical quantum mechanical methods that utilize a tight-binding approximation to density functional theory (DFT) [33]. They belong to a broader category of semiempirical methods that also includes the DFTB (Density Functional Tight-Binding) family [32]. The GFN framework encompasses several levels of theory, including GFN1-xTB, GFN2-xTB, GFN0-xTB, and the force-field method GFN-FF [33]. These methods have rapidly gained traction for efficient computational investigations across diverse chemical systems, from large transition-metal complexes to complex biomolecular assemblies [33].
GFN2-xTB: Capabilities and Limitations
GFN2-xTB has established itself as a valuable tool in computational chemistry, occupying "an odd niche" as noted in the literature [34]. It is described as "a parameterized tight-binding method that produces molecular geometries and noncovalent interactions fast enough for large-scale conformer searches, implicit solvent calculations, or molecular dynamics runs where DFT would be prohibitive" [34]. However, its limitations are well-documented: "reaction barriers come out too low, orbital gaps are compressed, and transition-metal complexes can sometimes distort into unphysical geometries" [34]. Despite these shortcomings, its speed and black-box nature maintain its popularity in many applications.
g-xTB: A General Replacement
The new g-xTB method from Grimme's group is explicitly intended as a general replacement for the GFN-xTB family [34]. The "g" stands for "general," and the development addresses three structural problems identified in GFN-xTB:
- No Hartree-Fock exchange: GFN2-xTB emulates a GGA functional, which works for geometries but is systematically weak for thermochemistry and barrier heights [34].
- Minimal basis sets: The traditional STO-nG-like bases don't adapt to molecular environments and lack polarization functions on key elements [34].
- Narrow training scope: Parameterization focused on a few property types rather than broad coverage of chemical space [34].
g-xTB maintains the tight-binding framework but introduces significant improvements including a charge-dependent, polarization-capable basis; an extended Hamiltonian with range-separated approximate Fock exchange; higher-order charge terms in the density-fluctuation expansion; atomic correction potentials; and a charge-dependent repulsion term [34].
Performance Comparison
The performance of electronic structure methods is typically evaluated using standardized benchmark sets like GMTKN55, which contains approximately 32,000 relative energies covering thermochemistry, kinetics, and noncovalent interactions [34]. On this benchmark, g-xTB demonstrates a weighted total mean absolute deviation (WTMAD-2) of 9.3 kcal/mol, roughly half that of GFN2-xTB and comparable to some "cheap" DFT methods [34]. This represents a substantial advancement in accuracy while maintaining the speed advantages of semiempirical methods.
Table 1: Overall Performance Comparison on GMTKN55 Benchmark
| Method | WTMAD-2 (kcal/mol) | Relative Performance |
|---|---|---|
| GFN2-xTB | ~18.6 | Baseline |
| g-xTB | 9.3 | ~50% improvement |
Bond Dissociation Energy (BDE) Prediction
For bond energy research—a critical aspect of the broader thesis on the accuracy of electronic structure methods—g-xTB shows remarkable improvement. On a recent set of experimental C-H and C-X bond dissociation energy benchmarks, g-xTB achieved a mean absolute error (MAE) of 3.96 kcal/mol after linear correction, far superior to GFN2-xTB's MAE of 7.88 kcal/mol [34]. This level of accuracy makes g-xTB comparable to top DFT methods or neural network potentials for this specific property [34]. The g-xTB//GFN2-xTB approach (single-point g-xTB calculations on GFN2-xTB geometries) emerged as one of the "two Pareto-dominant methods" and is recommended for production usage when efficiency is paramount [34].
Table 2: Bond Dissociation Energy Prediction Accuracy
| Method | Mean Absolute Error (kcal/mol) | Performance Note |
|---|---|---|
| GFN2-xTB | 7.88 | Significant scatter in predictions [34] |
| g-xTB | 3.96 | Strong correlation to experimental BDE [34] |
| g-xTB//GFN2-xTB | ~3.96 | Recommended for production use [34] |
Reaction Barrier Prediction
The inclusion of range-separated approximate Fock exchange in g-xTB directly addresses one of GFN2-xTB's key weaknesses: the systematic underestimation of reaction barriers. Literature confirms that "g-xTB is much better at predicting barrier heights" owing to this methodological improvement [34]. This advancement is particularly valuable for reaction mechanism studies and catalytic cycle analysis in high-throughput screening applications.
Geometrical and Electronic Properties
For geometry optimization of small organic semiconductor molecules, GFN2-xTB demonstrates high structural fidelity according to benchmarking studies [33]. However, g-xTB shows further improvements, particularly for transition-metal complexes, providing "more reliable and accurate geometries" [34]. For electronic properties, g-xTB "fixes the HOMO-LUMO gap issues exhibited by previous xTB methods and outperforms r2SCAN- or B3LYP-based methods at this task," a testament to the importance of range separation in the method [34].
Computational Efficiency
Computational efficiency is paramount for high-throughput screening applications. GFN2-xTB is recognized for its speed, enabling applications like conformer searches for transition-metal complexes through tools like CREST (Conformer-Rotamer Ensemble Sampling Tool) [35]. g-xTB maintains this practical advantage, being "only a little slower than GFN2-xTB (30% or less), making it essentially a drop-in replacement for any routine usage" [34]. It's important to note that the current release of g-xTB lacks analytical gradients, making geometry optimizations and frequency calculations significantly slower than GFN2-xTB [34].
Table 3: Computational Efficiency Comparison
| Method | Speed Relative to DFT | Key Considerations |
|---|---|---|
| GFN2-xTB | Orders of magnitude faster [34] | Fast geometry optimizations [35] |
| g-xTB | Similar to GFN2-xTB (≤30% slower) [34] | Lacks analytical gradients (slower optimizations) [34] |
Experimental Protocols and Workflows
High-Throughput Conformer Screening for Transition-Metal Complexes
The protocol below is adapted from studies analyzing Rh-based catalysts featuring bisphosphine ligands, which are widely employed in hydrogenation reactions [35]:
- Initial Conformer Generation: Perform conformer exploration using CREST (Conformer-Rotamer Ensemble Sampling Tool) with the GFN2-xTB//GFN-FF hybrid potential. This leverages the speed of GFN2-xTB for initial sampling [35].
- Conformer Filtering: Apply DBSCAN clustering using RMSD and energy parameters to eliminate redundancies while preserving key configurations. Studies show energy-based filtering is ineffective, while RMSD-based filtering improves selection [35].
- Geometry Refinement: Refine the filtered conformer ensembles via DFT geometry optimization using Gaussian 16 at the PBE0-D3(BJ)/def2-SVPP level of theory [35].
- Frequency Analysis: Confirm the nature of each stationary point via frequency analysis and compute thermochemical parameters at 298.15 K and 1 atm [35].
- Final Energy Evaluation: For production-level bond energies, perform single-point g-xTB calculations on the optimized GFN2-xTB or DFT geometries to leverage g-xTB's superior accuracy for bond dissociation energies [34].
Diagram 1: High-Throughput Conformer Screening Workflow
Protein-Ligand Interaction Energy Screening
For large systems beyond conventional DFT capabilities, such as protein-ligand interactions, the following protocol is recommended:
- System Preparation: Prepare protein and ligand structures, ensuring proper protonation states.
- Geometry Optimization: Optimize ligand geometry using GFN2-xTB (or g-xTB if analytical gradients become available).
- Interaction Energy Calculation: Perform single-point g-xTB calculations on the optimized structures to determine interaction energies. Benchmark results on the PLA15 dataset show g-xTB "outperforms other low-cost methods, including state-of-the-art neural network potentials" [34].
- Validation: For critical hits, validate with higher-level DFT calculations when computationally feasible.
Essential Computational Tools
Table 4: Research Reagent Solutions for Semi-Empirical Screening
| Tool/Reagent | Function | Application Note |
|---|---|---|
| CREST | Conformer-Rotamer Ensemble Sampling Tool for automated conformer search [35] | Uses GFN2-xTB//GFN-FF potential; overestimates ligand flexibility but efficient for initial sampling [35] |
| xTB Program | Main package for GFN-xTB calculations (GFN2-xTB and g-xTB) [35] | Current g-xTB release lacks analytical gradients [34] |
| GMTKN55 Database | Benchmark set with ~32,000 relative energies for method validation [34] | Contains thermochemistry, kinetics, and noncovalent interaction datasets [34] |
| DBSCAN Clustering | Density-based clustering algorithm for conformer filtering [35] | More effective than energy-based or PCA-based filtering; eliminates redundancies [35] |
| MORFEUS Python Package | Preprocessing and analysis of CREST output ensembles [35] | Facilitates filtering and analysis of conformer ensembles [35] |
The comparative analysis of GFN2-xTB and g-xTB reveals a clear trajectory of improvement in semiempirical quantum chemistry methods. While GFN2-xTB remains a valuable tool for rapid geometry optimization and initial conformer sampling—particularly when integrated into workflows like CREST—g-xTB represents a significant advancement in accuracy for key properties like bond dissociation energies, reaction barriers, and electronic properties.
For high-throughput screening applications in bond energy research, the evidence supports the following recommendations:
- For maximum throughput in geometry optimization and conformer sampling, GFN2-xTB remains the preferred choice.
- For accurate bond energy predictions and reaction barrier assessments, g-xTB single-point calculations on GFN2-xTB geometries provide an optimal balance of efficiency and accuracy.
- For protein-ligand interaction screening and other applications involving large systems, g-xTB offers superior performance compared to other low-cost methods.
The integration of these methods into computational screening pipelines demonstrates how semiempirical quantum chemistry continues to evolve, providing researchers with increasingly powerful tools for accelerating drug discovery and materials design while maintaining computational feasibility.
The accurate prediction of molecular properties, including bond dissociation energies (BDEs), is a cornerstone of computational chemistry, with profound implications for drug design, materials science, and energy storage research. The reliability of these predictions hinges on a foundational two-step workflow: geometry optimization to locate stable molecular structures on the potential energy surface, followed by single-point energy (SPE) calculations on these optimized geometries to compute highly accurate energies [36] [37]. This sequence balances computational cost with accuracy, as performing high-level theory calculations directly during geometry optimization is often prohibitively expensive for all but the smallest systems. The choice of methods for each step represents a series of trade-offs, creating a complex landscape that researchers must navigate. This guide provides an objective comparison of prevalent electronic structure methods, supported by experimental data, to inform best practices within the broader context of research on the accuracy of methods for calculating bond energies.
Method Comparison: Accuracy vs. Computational Cost
A hierarchical approach is widely recommended, where a lower-level (and faster) method is used for geometry optimization, and a higher-level (more accurate) method is used for the final single-point energy calculation [36] [37]. The following tables summarize the performance of various methods based on benchmarking studies.
Table 1: Comparison of Geometry Optimization Methods
| Method Class | Specific Methods | Typical Heavy-Atom RMSD (Å) vs. DFT | Relative Speed | Best Use Cases |
|---|---|---|---|---|
| Semi-empirical (GFN family) | GFN1-xTB, GFN2-xTB [33] | ~0.1 - 0.3 (for organic semiconductors) | 10⁴ - 10⁵ | High-throughput screening of organic molecules; pre-optimization [33]. |
| Density Functional Tight Binding (DFTB) | DFTB [36] | Similar to SEQM | ~10⁴ | Large systems where semi-empirical methods are insufficient. |
| Density Functional Theory (DFT) | PBE, B3LYP, r²SCAN-3c [37] [38] | Benchmark (0.0) | 1 - 100 (depends on functional/basis set) | Final, high-accuracy optimization; systems < 100 atoms [37]. |
| Neural Network Potentials (NNPs) | AIMNet2, OMol25 eSEN [39] | N/A (Data not provided in search results) | Varies (highly dependent on model and optimizer) | Promising as DFT replacements; performance depends heavily on optimizer choice [39]. |
Table 2: Performance of DFT Functionals for Single-Point Energy Calculations (for predicting redox potentials of quinones)
| DFT Functional | Level of Theory | RMSE (V) | R² | Recommended Use |
|---|---|---|---|---|
| PBE [36] | OPT(gas) + SPE(SOL) | 0.072 | 0.954 | Good balance of speed and accuracy for large-scale screening. |
| PBE [36] | OPT(gas) + SPE(SOL) | 0.051 | 0.977 | With implicit solvation, accuracy improves significantly. |
| B3LYP [36] | OPT(gas) + SPE(SOL) | ~0.055 | ~0.970 (estimated from graph) | Widely used; requires dispersion corrections for modern use [37]. |
| M08-HX [36] | OPT(gas) + SPE(SOL) | ~0.055 | ~0.970 (estimated from graph) | High-accuracy meta-GGA hybrid functional. |
| PBE0-D3 [36] | OPT(gas) + SPE(SOL) | ~0.050 (lowest error) | High | One of the most accurate functionals tested for redox potentials. |
Table 3: Optimizer Performance with Neural Network Potentials (Success rate for 25 drug-like molecules)
| Optimizer | OrbMol | OMol25 eSEN | AIMNet2 | Egret-1 | GFN2-xTB (Control) |
|---|---|---|---|---|---|
| ASE/L-BFGS | 22 | 23 | 25 | 23 | 24 |
| ASE/FIRE | 20 | 20 | 25 | 20 | 15 |
| Sella (internal) | 20 | 25 | 25 | 22 | 25 |
| geomeTRIC (tric) | 1 | 20 | 14 | 1 | 25 |
Experimental Protocols: Detailed Methodologies for Key Workflows
A Standard Protocol for High-Throughput Screening
A systematic workflow for the discovery of quinone-based electroactive compounds demonstrates a robust hierarchical protocol [36].
- Initial Structure Generation: The molecular structure begins as a SMILES string, which is converted to a 3D geometry.
- Geometry Optimization: The 3D structure is optimized using a force field (e.g., OPLS3e) or a fast semi-empirical method (e.g., GFN-xTB) to locate a reasonable low-energy conformation.
- Higher-Level Optimization (Optional): The geometry may be refined further using a more accurate method, such as DFT. Notably, the study found that optimizing geometries in the gas phase, rather than with an implicit solvation model, was sufficient and computationally cheaper [36].
- Single-Point Energy Calculation: The key energetic properties are calculated by performing a single-point energy calculation on the optimized geometry using a higher-level DFT functional (e.g., PBE0-D3) and including an implicit solvation model (e.g., Poisson-Boltzmann) to account for solvent effects.
- Property Prediction: The reaction energy (ΔEᵣₓₙ) computed from the SPE is then calibrated against experimental data (e.g., redox potentials) to validate the protocol.
Achieving Chemical Accuracy for Bond Dissociation Energies (BDEs)
For calculating accurate BDEs of challenging molecules like per- and polyfluoroalkyl substances (PFAS), a fragmentation-based protocol can overcome the limitations of standard DFT [40].
- Initial Geometry Optimization: The parent molecule and its dissociation fragments are optimized at the DFT level (e.g., using M062X/6-31G(2df,p)).
- Connectivity-Based Hierarchy (CBH): The parent molecule is divided into small, well-defined fragments based on its molecular connectivity. The "modified CBH" scheme, which saturates carbon valences with fluorine atoms for PFAS, is recommended to preserve the chemical environment [40].
- High-Level Energy Calculation: The ground-state free energies (GSFEs) of these small fragments are computed at a high-level composite method (e.g., G4 theory).
- Energy Reconstruction: The accurate GSFE of the large parent molecule is estimated by summing the DFT energy of the parent and the correlation energy corrections (difference between G4 and DFT energies) of all fragments.
- BDE Computation: The BDE is calculated as the difference between the accurately estimated GSFEs of the dissociated fragments and the parent molecule. This approach has been shown to achieve near-chemical accuracy (< 1 kcal mol⁻¹) at a computational cost comparable to a standard DFT calculation [40].
Workflow Visualization
The following diagram illustrates the standard multi-level computational workflow for going from a molecular structure to accurate energy-based properties, integrating the key decision points and protocols discussed.
The Scientist's Toolkit: Essential Computational Reagents
Table 4: Key Software and Method Components for the Computational Workflow
| Tool Name | Type | Primary Function | Considerations |
|---|---|---|---|
| GFN-xTB [33] | Semi-empirical Method | Fast geometry optimization and pre-screening for organic molecules. | GFN1/2-xTB offer high structural fidelity; GFN-FF provides the best speed/accuracy balance for large systems. |
| DFT Functionals (PBE0-D3, r²SCAN-3c) [36] [37] | Quantum Chemical Method | Accurate single-point energy and electronic property calculations. | r²SCAN-3c is a robust, modern composite method. PBE0-D3 shows high accuracy for redox properties. |
| B3LYP-D3 [37] [40] | Quantum Chemical Method | A widely used functional, now requiring dispersion corrections (D3) for reliable results. | Outperforms plain B3LYP; when combined with mCBH scheme, can yield accurate BDEs. |
| Sella [39] | Geometry Optimizer | Optimizes structures to minima or transition states using internal coordinates. | Shows excellent performance and step efficiency with various NNPs and GFN2-xTB. |
| geomeTRIC [39] | Geometry Optimizer | General-purpose optimization library using translation-rotation internal coordinates (TRIC). | Performance is highly dependent on the coordinate system and the NNP used. |
| Connectivity-Based Hierarchy (CBH) [40] | Fragmentation Scheme | Achieves near-chemical accuracy for energies of large molecules at DFT cost. | Essential for reliable BDEs of large systems like PFAS; the "modified CBH" adapts to the chemical environment. |
| Implicit Solvation Models (e.g., PBF) [36] | Solvation Method | Models solvent effects on molecular geometry and energy in a computationally efficient way. | Critical for predicting properties in solution; inclusion in SPE calculations significantly improves accuracy. |
Optimizing for Accuracy and Efficiency: Strategies for Robust Bond Energy Calculations
The selection of a basis set is a fundamental step in quantum chemical calculations, representing a critical trade-off between computational cost and accuracy. [41] This guide provides an objective comparison of two basis sets—the double-zeta polarized vDZP and the triple-zeta polarized def2-TZVPPD—within the specific context of predicting bond dissociation energies (BDEs), a property vital for understanding reactivity and stability in drug development.
Basis sets form the mathematical basis for expanding molecular orbitals, with their size and quality directly impacting the description of the electron cloud. [41] The hierarchy progresses from minimal single-zeta (SZ) to double-zeta (DZ), triple-zeta (TZ), and quadruple-zeta (QZ) basis sets, with the addition of polarization (P) and diffuse (D) functions crucial for accurately capturing electron distribution and non-covalent interactions. [42] While conventional wisdom often recommends triple-zeta basis sets for high-quality results, their computational cost can be prohibitive for large systems, prompting the development of efficient alternatives like vDZP. [43]
Basis Set Comparison: vDZP vs. def2-TZVPPD
The table below summarizes the key characteristics of the vDZP and def2-TZVPPD basis sets.
Table 1: Fundamental Characteristics of the Basis Sets
| Basis Set | Zeta (ζ) Quality | Key Features | Intended Use Case |
|---|---|---|---|
| vDZP [43] [15] | Double-Zeta | Uses effective core potentials and deeply contracted valence functions; designed to minimize BSSE. | Rapid, efficient calculations on large systems where triple-zeta costs are prohibitive. |
| def2-TZVPPD [15] [44] | Triple-Zeta | Triple-zeta valence with a double set of polarization functions and added diffuse functions. | High-accuracy energy calculations, especially where electron affinity or non-covalent interactions are important. |
Performance Comparison for Bond Dissociation Energies
Quantitative Accuracy Assessment
The performance of these basis sets was evaluated using the ExpBDE54 benchmark, a dataset of 54 experimental homolytic bond-dissociation enthalpies for carbon-hydrogen and carbon-halogen bonds. [15] The electronic bond-dissociation energy (eBDE) was calculated using the highly accurate r2SCAN-D4 functional combined with each basis set. A linear regression correction was then applied to the eBDEs to account for the lack of zero-point energy, enthalpy, and relativistic effects, enabling a direct comparison with experimental values. [15]
Table 2: Performance on the ExpBDE54 Benchmark for BDE Prediction
| Method | Basis Set | Root-Mean-Square Error (RMSE) | Relative Computational Speed |
|---|---|---|---|
| r2SCAN-D4 [15] | def2-TZVPPD | 3.6 kcal/mol | 1.0x (Reference) |
| r2SCAN-D4 [15] | vDZP | ~5.1 kcal/mol | ~2.0x faster |
| r2SCAN-D4 [15] | def2-QZVP | ~3.6 kcal/mol | ~1.9x slower |
The data shows that def2-TZVPPD achieves higher accuracy, with an RMSE close to the likely basis set limit as indicated by the def2-QZVP result. [15] However, vDZP offers a significant speed advantage, being about twice as fast as def2-TZVPPD with a moderate increase in error. [15]
Performance Across Different Functionals
The robustness of a basis set is measured by its ability to deliver good performance across a wide range of density functionals without need for reparameterization. The vDZP basis set has been tested extensively with various functionals on the comprehensive GMTKN55 thermochemistry benchmark. [43]
Table 3: vDZP Performance with Different Density Functionals (WTMAD2 from GMTKN55)
| Functional | Basis Set | Overall WTMAD2 | Performance Summary |
|---|---|---|---|
| B97-D3BJ [43] | def2-QZVP | 8.42 | Reference accuracy with a large basis set. |
| B97-D3BJ [43] | vDZP | 9.56 | Good performance, slightly reduced accuracy. |
| r2SCAN-D4 [43] | def2-QZVP | 7.45 | Reference accuracy with a large basis set. |
| r2SCAN-D4 [43] | vDZP | 8.34 | Good performance, slightly reduced accuracy. |
The results demonstrate that vDZP provides consistent and robust performance across multiple functional types, including GGA (B97-D3BJ), meta-GGA (r2SCAN-D4), hybrid (B3LYP-D4), and hybrid meta-GGA (M06-2X). [43] The moderate increase in error compared to large basis-set references is consistent, making it a reliable and efficient choice.
Experimental Protocols for Basis Set Benchmarking
Workflow for Energy and Property Calculation
The following diagram illustrates a standardized computational workflow for benchmarking basis sets, as employed in the cited studies. [15] [45]
Detailed Methodology for BDE Calculation
The protocol for calculating bond dissociation energies, as used in the ExpBDE54 benchmark, involves: [15]
- Initial Structure Generation: Molecular structures are generated from SMILES strings and undergo an initial geometry optimization using the GFN2-xTB semiempirical method to provide a reasonable starting geometry.
- Target-Method Optimization: The structure is re-optimized using the target density functional (e.g., r2SCAN-D4) and the target basis set (e.g., vDZP or def2-TZVPPD).
- Bond Cleavage and Fragment Optimization: The bond of interest is cleaved homolytically, generating two doublet radical fragments. These fragments are then geometrically optimized using the same method.
- Electronic Energy Calculation: The electronic bond dissociation energy (eBDE) is calculated as the difference between the sum of the electronic energies of the optimized fragments and the electronic energy of the optimized parent molecule:
eBDE = E(fragment1) + E(fragment2) - E(parent). - Correction and Validation: A linear regression model is fitted to correct the calculated eBDE values against experimental BDEs. This step accounts for systematic errors and missing terms like zero-point energy. The root-mean-square error (RMSE) between the corrected calculated values and experimental values is the primary metric for accuracy.
The Scientist's Toolkit: Key Computational Reagents
Table 4: Essential Software and Methods for Computational Chemistry
| Tool / Method | Function | Relevance to Basis Set Studies |
|---|---|---|
| Psi4 [43] [15] | A suite of ab initio quantum chemistry programs. | The primary software used for running DFT calculations with different functionals and basis sets in the cited benchmarks. |
| geomeTRIC [43] [45] | A geometry optimization package. | Used to perform stable and efficient geometry optimizations during the computational workflow. |
| GFN2-xTB [15] [45] | A semiempirical quantum mechanical method. | Often used for fast initial geometry optimizations and conformational searching to provide input structures for more expensive DFT calculations. |
| GMTKN55 [43] | A comprehensive database of 55 benchmark sets for main-group chemistry. | Used for the broad evaluation of method performance across diverse chemical properties like thermochemistry and non-covalent interactions. |
| ExpBDE54 [15] | A curated set of 54 experimental Bond Dissociation Enthalpies. | Provides a slim, focused benchmark for validating the accuracy of computational methods for predicting bond strengths. |
| D3BJ & D4 Dispersion [43] [15] | Empirical corrections for dispersion interactions. | Crucial additions to density functionals for accurately describing van der Waals forces and non-covalent interactions in conjunction with the basis set. |
The choice between the vDZP and def2-TZVPPD basis sets is a direct reflection of the accuracy-cost paradigm in computational chemistry. For high-accuracy prediction of bond dissociation energies and other electronic properties, def2-TZVPPD is the superior choice, providing results near the basis set limit. However, for researchers studying large systems, screening molecular libraries, or where computational throughput is paramount, vDZP offers an excellent compromise, delivering robust and respectable accuracy at approximately double the speed. Its consistent performance across multiple density functionals makes it a versatile and powerful tool for the modern computational scientist.
Addressing Open-Shell Systems and Overcoming SCF Convergence Challenges
Electronic structure calculations for open-shell systems—those with unpaired electrons—present unique challenges for quantum chemistry methods. Accurately describing these systems is crucial across many chemical domains, from understanding transition metal catalysts in drug development to characterizing magnetic materials and radical reaction mechanisms. The inherent complexity arises from the need to properly account for spin polarization and electron correlation while maintaining manageable computational cost.
The Self-Consistent Field (SCF) procedure, fundamental to both Hartree-Fock and Density Functional Theory (DFT) calculations, often encounters convergence difficulties when applied to open-shell systems. These challenges stem from several factors: the presence of near-degenerate electronic states, complex spin coupling scenarios, and the limitations of initial guess orbitals. When SCF convergence fails, researchers may obtain unrealistic electronic configurations, higher-energy excited states, or no solution at all, compromising the reliability of subsequent property predictions [46].
This guide objectively compares the performance of various electronic structure methods for addressing open-shell systems and SCF convergence challenges, with particular emphasis on their accuracy for calculating bond energies—a critical property in drug development and materials science.
Methodological Approaches for Open-Shell Systems
Comparative Analysis of Electronic Structure Methods
Table 1: Comparison of Electronic Structure Methods for Open-Shell Systems
| Method | Spin Symmetry | Computational Cost | Key Strengths | Key Limitations | Typical Bond Energy RMSE (kcal/mol) |
|---|---|---|---|---|---|
| UHF/UKS | Not conserved (spin contamination) | Low | Black-box implementation, describes spin polarization | Spin contamination, unreliable for complex spin coupling | 4-6 (highly functional-dependent) [15] |
| ROHF/ROKS | Conserved (spin eigenfunction) | Low to moderate | Spin-pure wavefunction, better starting point for correlated methods | Poorer convergence, limited to high-spin cases in basic implementations | 3.6-4.7 (when properly corrected) [15] |
| g-ROHF | Conserved (spin eigenfunction) | Moderate | Handles arbitrary spin couplings, supports orbital degeneracies | Requires specialized implementation, less widely available | Methodology under active development [47] |
| CASSCF | Conserved (spin eigenfunction) | Very high to prohibitive | Handles complex spin coupling, multi-reference character | Exponential scaling with active space size, expensive | Potential for high accuracy but system-dependent [47] |
| CCSD(T) | Conserved (when based on ROHF) | Extremely high | "Gold standard" for accuracy, reliable for correlation effects | Limited to small systems (~10 atoms), poor scaling | ~1-2 (but limited to small systems) [5] |
| NEVPT2 | Conserved (when based on CASSCF) | High | Accounts for dynamic correlation, good for transition metals | Depends on quality of active space selection | System-dependent, generally more reliable than DFT [48] |
For researchers requiring rapid screening of bond dissociation enthalpies (BDEs), recent benchmarks on the ExpBDE54 dataset reveal that suitably corrected semiempirical and machine-learning approaches can offer compelling performance. The g-xTB//GFN2-xTB and OMol25's eSEN Conserving Small methods define the Pareto frontier for this application, yielding root-mean-square errors of 4.7 and 3.6 kcal·mol⁻¹ respectively, while offering substantial computational savings over traditional DFT [15].
Specialized Techniques for Complex Systems
For systems exhibiting particularly challenging electronic structures, such as transition metal complexes with significant multiconfigurational character or metal-radical assemblies, specialized methodologies have emerged. The general Restricted Open-Shell Hartree-Fock (g-ROHF) approach represents a compelling middle ground between the structural shortcomings of UHF and the computational demands of CASSCF [47]. Unlike traditional ROHF methods typically restricted to high-spin cases, the g-ROHF formulation supports general-spin couplings and orbital degeneracies while preserving spin purity, making it particularly valuable for calculating properties like g-tensors and hyperfine couplings in experimentally characterized systems such as mixed-valence manganese(III/IV) dimers and the metal-radical complex Fe(GMA)(pyridine)⁺ [47].
For the most challenging systems where even single-reference methods fail, multi-configurational approaches become necessary. The spin-flip methodology provides an elegant alternative, starting from a single determinant of the high-spin type where all unpaired electrons are aligned in parallel, then applying spin-flip operators to access lower multiplicities and more complex spin-coupling situations [47].
Protocols for SCF Convergence Enhancement
Standard Convergence Protocols
Achieving SCF convergence in open-shell systems often requires specialized techniques beyond default settings. The following workflow outlines a systematic approach to addressing convergence challenges:
For spin-unrestricted calculations, the initial guess can be crucial. The ADF documentation recommends using the MODIFYSTARTPOTENTIAL keyword or the SPINFLIP option in the RESTART key to start calculations in broken symmetry, for example with spin-α density on one fragment and spin-β density on another [46]. This approach is particularly valuable for studying systems like separated H₂ molecules at large distances where proper spin localization is essential.
When these standard approaches prove insufficient, more advanced strategies are necessary. For systems with spin-orbit coupling, different convergence protocols apply. In these cases, researchers should use Unrestricted Yes with Symmetry NOSYM and select either collinear or noncollinear approximation within the relativity block [46]. The SpinOrbitMagnetization key can be employed to define starting spin-polarization directions for specific atomic regions, providing finer control over the initial guess [46].
Advanced and System-Specific Protocols
For particularly challenging systems, advanced SCF protocols offer additional avenues for achieving convergence:
Restricted Open-Shell SCF (ROSCF): ADF2023 implemented a restricted open shell method valid for high-spin open-shell molecules, where the one-determinantal wave function is an eigenfunction of S₂ and S² [46]. The input structure requires integer occupation numbers, a positive spin polarization, and the ROSCF subkey:
This method currently applies primarily to single-point calculations but demonstrates improved stability for appropriate systems [46].
Spin-Orbit Coupled Calculations: For systems requiring explicit spin-orbit coupling treatment, specialized protocols are essential. The collinear approximation can be implemented with:
While the noncollinear approximation uses:
In these cases, traditional convergence keys like MODIFYSTARTPOTENTIAL and RESTART%spinflip cannot be used, and the SpinOrbitMagnetization key should be employed instead [46].
Performance Assessment for Bond Energy Calculations
Benchmarking Methodologies
Accurately assessing the performance of electronic structure methods for bond energy predictions requires carefully designed benchmark studies. The ExpBDE54 dataset provides a valuable resource for this purpose, comprising experimental gas-phase bond dissociation enthalpies for 54 small molecules covering carbon-hydrogen and carbon-halogen bonds—motifs particularly relevant to organic and medicinal chemistry [15].
The standard protocol for calculating bond dissociation energies involves several methodical steps:
Initial Structure Generation: Molecular structures are generated from SMILES strings and pre-optimized with a low-cost method like GFN2-xTB to provide a consistent starting point for all subsequent calculations [15].
Electronic Bond Dissociation Energy (eBDE) Calculation: The initial structure is optimized with the target method, then the target bond is cleaved homolytically to create two doublet fragments. Fragments with more than one atom are optimized, and the eBDE is calculated as the electronic energy difference between the molecule and its fragments [15].
Thermochemical Correction: A linear regression is fit to the eBDEs relative to experimental bond-dissociation enthalpies to correct for the lack of zero-point energy, enthalpy, and relativistic effects [15].
This methodology ensures consistent comparison across methods while acknowledging that purely electronic energy differences require empirical correction to match experimental enthalpies.
Comparative Performance Data
Table 2: Accuracy and Efficiency of BDE Prediction Methods (ExpBDE54 Benchmark)
| Method Class | Specific Method | RMSE (kcal/mol) | Relative Speed | Recommended Use Case |
|---|---|---|---|---|
| Semiempirical | GFN2-xTB | ~5.0 | 1000x | Initial screening, large systems |
| Semiempirical | g-xTB//GFN2-xTB | 4.7 | 500x | Rapid screening with moderate accuracy |
| Machine Learning | OMol25 eSEN Conserving Small | 3.6 | 1000x | Medium-sized systems, high throughput |
| meta-GGA DFT | r2SCAN-3c//GFN2-xTB | ~4.0 | 100x | Best speed/accuracy tradeoff for QM |
| Hybrid DFT | ωB97M-D3BJ/def2-TZVPPD | 3.7 | 10x | High accuracy for medium systems |
| Double-Hybrid DFT | DSD-BLYP-D3BJ/def2-TZVPPD | ~3.8 | 1x | High accuracy for small systems |
The benchmark data reveals several important trends. First, suitably corrected semiempirical and machine-learning approaches can achieve accuracy competitive with many DFT functionals while offering order-of-magnitude speed improvements [15]. The Pareto frontier for this application is currently defined by g-xTB//GFN2-xTB and OMol25's eSEN Conserving Small, demonstrating that modern approximate methods have matured significantly [15].
For DFT-based approaches, the r2SCAN-D4/def2-TZVPPD combination outperforms other functional/basis set combinations in this benchmark, with a linear correction yielding an RMSE of 3.6 kcal/mol with respect to experimental BDEs [15]. Moving to larger basis sets like def2-QZVP provided negligible improvement in accuracy while increasing computational time by 1.9x, suggesting the basis set limit for BDE prediction had essentially been reached [15].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Open-Shell Systems
| Tool Name | Type | Primary Function | Open-Shell Specialization |
|---|---|---|---|
| ADF | Software Suite | DFT calculations with specialized relativity treatment | Advanced open-shell methods, spin-orbit coupling, ROSCF implementation [46] |
| ORCA | Software Suite | Ab initio quantum chemistry | General ROHF (g-ROHF) implementation, EPR property calculations [47] |
| PySCF | Python Package | Quantum chemistry | Generalized Hartree-Fock (GHF) for noncollinear spins [49] |
| DIRAC | Software Suite | Relativistic quantum chemistry | Four-component calculations, Kramers-restricted methods [49] |
| FreeQuantum | Computational Pipeline | Binding energy calculations | Quantum computing-ready framework for challenging systems [48] |
| AutoSolvateWeb | Web Platform | Solvation environment modeling | Explicit solvent effects for open-shell systems [50] |
| ExpBDE54 | Benchmark Dataset | Method validation | Experimental BDEs for accuracy assessment [15] |
Specialized software tools are essential for addressing the unique challenges of open-shell systems. The ADF software suite provides comprehensive functionality for open-shell systems, including unrestricted calculations, spin-orbit coupling treatments, and the recently implemented ROSCF method for high-spin open-shell molecules [46]. ORCA has featured general ROHF capabilities since its early development, providing a robust platform for treating complex open-shell molecules and serving as the foundation for methods like ROCIS (Restricted Open-Shell Configuration Interaction Singles) for spectroscopic applications [47].
For modeling explicitly solvated open-shell systems—particularly relevant for drug development applications—cloud-based platforms like AutoSolvateWeb offer accessible solutions. This chatbot-assisted platform guides users through multistep procedures involving various computational packages to configure and execute quantum mechanical/molecular mechanical (QM/MM) simulations of explicitly solvated molecules, eliminating hardware configuration barriers for non-specialists [50].
Emerging computational pipelines like FreeQuantum represent the cutting edge, integrating machine learning, classical simulation, and high-accuracy quantum chemistry in a modular system designed to eventually incorporate quantum computing for the most computationally intensive subproblems [48]. Tested on a ruthenium-based anticancer drug, this framework produced significantly different binding energy predictions than classical methods, underscoring the value of quantum-level accuracy for pharmaceutically relevant systems [48].
The accurate prediction of bond-dissociation enthalpies (BDEs) is a cornerstone of computational chemistry, with critical applications in predicting the rates and regioselectivity of free-radical reactions, as well as identifying potential sites of drug metabolism [15]. However, computationally derived electronic bond-dissociation energies (eBDEs) represent differences in electronic energy at 0 K and lack the thermodynamic contributions present in experimental BDE measurements. To bridge this gap, linear regression corrections have emerged as a vital post-processing technique. This method uses a simple statistical model to account for systematic errors and missing enthalpic contributions, enabling computationally efficient methods to achieve accuracy comparable to more expensive approaches. This guide objectively compares the performance of various electronic structure methods when enhanced with linear regression corrections, providing researchers with a framework for selecting appropriate computational workflows for BDE prediction.
Theoretical Background and Methodological Rationale
The Need for Corrections in Electronic Structure Calculations
Pure electronic structure calculations, while providing the foundation for understanding chemical bonding, inherently lack several physical components necessary for direct comparison with experimental thermodynamic data:
- Zero-Point Energy (ZPE): The vibrational energy present at absolute zero is not included in the electronic energy.
- Thermal Enthalpic Contributions: Experimental BDEs are typically measured at 298 K and include energy from populated vibrational, rotational, and translational modes.
- Relativistic Effects: For heavier elements, these can become non-negligible [15].
Attempting to calculate these terms explicitly for large systems, through frequency calculations for instance, becomes computationally prohibitive. Linear regression offers a practical and efficient alternative. By establishing a linear relationship between computed eBDEs and experimental BDEs for a benchmark set, a correction is derived that implicitly accounts for the missing physical effects. This approach is particularly powerful because it can also correct for systematic errors inherent to the specific electronic structure method used.
The Linear Regression Workflow
The general protocol for applying linear regression corrections to BDE prediction is illustrated below and involves a clear sequence of computational and statistical steps.
Diagram 1: Workflow for BDE Prediction Using Linear Regression Correction. The pre-established calibration phase (dashed box) is performed once for a given computational method and benchmark set.
Experimental Protocols and Benchmarking
The ExpBDE54 Benchmark Set
A critical component of this methodology is the use of a reliable benchmark set. The ExpBDE54 set is a "slim" experimental benchmark comprising 54 small molecules, primarily featuring carbon-hydrogen and carbon-halogen bonds, which are highly relevant to organic and medicinal chemistry [15]. This set was compiled from carefully curated experimental gas-phase BDE measurements [15]. Its purpose is not for training machine-learning models but to serve as an external validation set for assessing the real-world performance of different computational workflows.
Detailed Computational Methodology
The following protocols are adapted from the ExpBDE54 benchmarking study [15] and represent standard practices in the field.
Protocol 1: Workflow for DFT-based BDE Prediction
- Initial Structure Generation: Generate a 3D molecular structure from a SMILES string.
- Geometry Optimization: Optimize the molecular geometry using a cost-effective method like GFN2-xTB to provide a reliable starting structure for all subsequent calculations.
- Single-Point Energy Calculation: Calculate the high-precision electronic energy of the optimized structure using the chosen Density Functional Theory (DFT) functional and basis set.
- Bond Cleavage and Fragment Optimization: Homolytically cleave the target bond. For fragments with more than one atom, perform a geometry optimization of the resulting doublet radicals.
- Electronic BDE (eBDE) Calculation: Compute the eBDE as the difference between the sum of the electronic energies of the optimized fragments and the electronic energy of the parent molecule:
eBDE = E(fragment A) + E(fragment B) - E(parent molecule). - Linear Regression Correction: Apply a pre-determined linear correction (
BDE_corrected = Slope × eBDE + Intercept) to account for ZPE, enthalpic, and systematic errors.
Protocol 2: Workflow for Semiempirical and Machine Learning Methods
- Initial Structure Generation: As in Protocol 1.
- Geometry Optimization: Optimize the structure with GFN2-xTB.
- Energy Evaluation: Calculate the electronic energy using a fast semiempirical method (e.g., GFN1-xTB, g-xTB) or a neural network potential (NNP) like OMol25's eSEN. Note: For some methods like the preliminary g-xTB, a single-point energy calculation on the GFN2-xTB-optimized geometry is performed if analytical gradients are unavailable.
- Fragment Energy Calculation: Perform energy calculations (with optimization if required) on the radical fragments generated by homolytic bond cleavage.
- eBDE and Correction: Calculate the eBDE and apply the method-specific linear regression correction.
Performance Comparison of Electronic Structure Methods
The performance of a computational method is typically evaluated by its Root-Mean-Square Error (RMSE) against the experimental benchmark, which measures the average magnitude of prediction errors. The ideal method has a low RMSE and low computational cost.
Table 1: Performance of Electronic Structure Methods with Linear Regression Corrections on the ExpBDE54 Set [15]
| Method Class | Specific Method | Basis Set / Details | RMSE (kcal·mol⁻¹) | Relative Speed (vs. r2SCAN-D4/TZVPPD) | Key Application Context |
|---|---|---|---|---|---|
| Semiempirical | g-xTB//GFN2-xTB [15] | N/A | 4.7 | ~100x Faster | CPU-based high-throughput screening; best speed/accuracy on CPU. |
| Neural Network Potential | OMol25 eSEN Conserving Small [15] | N/A | 3.6 | Varies | Medium-sized systems; defines Pareto frontier for accuracy. |
| Meta-GGA DFT | r2SCAN-3c//GFN2-xTB [15] | mTZVPP | ~4.0 | ~2.5x Faster | Best speed/accuracy trade-off for a QM-based method. |
| Meta-GGA DFT | r2SCAN-D4 [15] | def2-TZVPPD | 3.6 | 1.0 (Reference) | High-accuracy reference; approaching limit of electronic energy accuracy. |
| Meta-GGA DFT | r2SCAN-D4 [15] | def2-QZVP | ~3.6 | ~0.5x Slower | Negligible gain over TZVPPD; can have SCF convergence issues. |
| Hybrid DFT | B3LYP-D4 [15] | def2-TZVPPD | 4.1 | ~2x Faster | Good performance but less accurate than r2SCAN-D4. |
Table 2: Performance Comparison of Different Basis Sets with r2SCAN-D4 Functional [15]
| Basis Set | Zeta (ζ) Quality | RMSE (kcal·mol⁻¹) | Relative Speed | Comment |
|---|---|---|---|---|
| vDZP [15] | 2 | ~5.1 | ~2x Faster | Good for initial scans; increased RMSE. |
| def2-TZVPPD [15] | 3 | 3.6 | 1.0 (Reference) | Sweet spot for BDE prediction; likely at basis set limit. |
| def2-QZVP [15] | 4 | ~3.6 | ~1.9x Slower | No meaningful improvement over TZVPPD; not cost-effective. |
Analysis of Comparative Performance
The data reveals several key trends for researchers:
- The Pareto Frontier: The semiempirical g-xTB//GFN2-xTB and the neural network potential OMol25's eSEN define the "Pareto frontier," meaning they offer the best possible compromise between speed and accuracy—no other method is both faster and more accurate than these two for this specific task [15].
- Basis Set Saturation: For BDE prediction, moving from a triple-zeta (3ζ) basis set (e.g., def2-TZVPPD) to a quadruple-zeta (4ζ) set offers no significant improvement in accuracy but nearly doubles the computational cost [15]. This indicates that the basis set limit for BDE prediction is effectively reached with high-quality 3ζ sets.
- Composite Methods are Efficient: The composite method r2SCAN-3c, which uses a specialized basis set and integrated dispersion correction, provides excellent accuracy (RMSE ~4.0 kcal·mol⁻¹) at a significantly reduced cost, making it a highly recommended general-purpose tool [15].
- Accuracy Limit: The study suggests that with linear regression, these methods are approaching the limit of accuracy achievable using only the electronic energy. Further improvements would require explicit enthalpy calculations or group-specific corrections [15].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools for BDE Prediction
| Tool / Solution | Function / Description | Relevance to BDE Prediction |
|---|---|---|
| GFN2-xTB | A semiempirical method for fast geometry optimization [15]. | Provides reliable starting structures for higher-level single-point energy calculations, reducing overall computational time. |
| r2SCAN-3c | A cost-effective, composite DFT method with a tailored basis set and dispersion correction [15]. | An excellent balance of speed and accuracy for routine BDE predictions on medium-sized systems. |
| g-xTB | A GPU-accelerated semiempirical quantum chemistry method [15]. | Enables high-throughput BDE screening of large molecular libraries by drastically reducing computation time. |
| OMol25 eSEN | A Neural Network Potential (NNP) trained on quantum chemical data [15]. | Offers high accuracy (near-DFT) at a fraction of the cost; ideal for projects with sufficient training data. |
| def2-TZVPPD | A high-quality triple-zeta basis set with diffuse functions [15]. | The recommended basis set for achieving chemical accuracy in DFT-based BDE calculations without excessive cost. |
| Linear Regression Parameters | The slope and intercept derived from benchmarking [15]. | The essential correction that transforms raw electronic energies into accurate, experimentally comparable BDEs. |
| ExpBDE54 Dataset | A curated set of 54 experimental BDEs for small molecules [15]. | Serves as a benchmark for validating and calibrating new computational workflows. |
The application of linear regression corrections represents a powerful and pragmatic approach to achieving high accuracy in BDE predictions from electronic structure methods. The comparative data clearly shows that modern semiempirical and machine-learning methods, when properly corrected, can compete with the accuracy of more expensive DFT methods while offering order-of-magnitude speedups.
For the researcher, the choice of method depends on the specific project goals:
- For high-throughput virtual screening in drug discovery, where speed is paramount, g-xTB//GFN2-xTB is the leading candidate.
- For the most accurate studies on medium-sized systems where computational resources are available, r2SCAN-D4/def2-TZVPPD is an excellent choice.
- For the best balance of speed and accuracy in a traditional quantum mechanics method, r2SCAN-3c//GFN2-xTB is highly recommended.
Future developments will likely focus on refining group-specific corrections and integrating more sophisticated machine-learning models that can capture non-linear relationships, potentially pushing accuracy beyond the current limits of linear models. The continued development of efficient, GPU-accelerated codes and accurate neural network potentials will further expand the scope of molecules and properties accessible to rapid computational prediction.
In the realm of computational chemistry, researchers constantly face a fundamental challenge: the trade-off between the computational cost of a calculation and the accuracy of its results. This is particularly crucial in drug design, where predicting the binding affinity of ligands to protein pockets can significantly accelerate the early stages of the drug development pipeline [51]. The Pareto frontier is a powerful conceptual framework that helps navigate this trade-off. It represents the set of optimal configurations where no further improvement can be made in one metric (e.g., speed) without worsening the other (e.g., accuracy) [52]. For a fixed model and task configuration, constructing a Pareto frontier allows researchers to make critical claims, such as determining the minimum computational time required to achieve a desired accuracy for a binding energy calculation, or identifying the best possible accuracy attainable within a fixed computational budget [52].
Understanding and applying this concept is indispensable. Inaccuracies as small as 1 kcal/mol in binding energy predictions can lead to erroneous conclusions about relative binding affinities, potentially derailing a drug discovery project [51]. This article provides a comparative guide to the performance of various electronic structure methods, arming researchers with the data needed to identify their own optimal Pareto frontier for projects involving the calculation of bond dissociation energies and other non-covalent interactions.
Theoretical Framework: Speed-Accuracy Trade-offs
The speed-accuracy tradeoff (SAT) is a ubiquitous phenomenon observed across biological and artificial decision-making systems [53] [54]. In cognitive psychology, it describes the tendency for decision speed to covary with decision accuracy; faster decisions typically come at the cost of higher error rates, and vice versa [53] [55]. This concept translates directly to computational chemistry. Here, the "decision" is the output of an electronic structure calculation, such as a bond length or energy. The "speed" is the computational time and resources required. The "accuracy" is the deviation of the calculated result from a reliable experimental or benchmark theoretical value.
The neurophysiological underpinnings of SAT in humans provide an interesting analogy for computational systems. The threshold hypothesis posits that SAT is managed by adjusting the amount of evidence (information) required to make a decision [54]. In computational terms, this is akin to a method's level of approximation; a "fast" method uses a lower threshold of information (e.g., a smaller basis set, a simpler functional) to reach a result quickly, while an "accurate" method integrates more information (e.g., a larger basis set, a higher level of theory) at a greater computational cost [54]. This framework allows us to rationally compare and categorize different computational approaches.
Benchmarking Electronic Structure Methods for Bond Energies
The Quest for a "Platinum Standard" in Benchmarking
Robust quantum-mechanical (QM) benchmarks are essential for evaluating the performance of electronic structure methods, but they are scarce for large, biologically relevant ligand-pocket systems [51]. Disagreements between established "gold standard" methods like Coupled Cluster (CC) and Quantum Monte Carlo (QMC) have further complicated the benchmarking landscape for larger non-covalent systems [51]. To address this, the "QUantum Interacting Dimer" (QUID) benchmark framework was recently introduced. It contains 170 non-covalent systems modeling chemically and structurally diverse ligand-pocket motifs [51]. A key innovation of QUID is the establishment of a "platinum standard," achieved by securing tight agreement (0.5 kcal/mol) between two fundamentally different high-level methods: LNO-CCSD(T) and FN-DMC [51]. This significantly reduces the uncertainty in the highest-level QM calculations and provides a reliable yardstick for assessing more approximate methods.
Comparative Performance of Computational Methods
Table 1: Performance of Select Electronic Structure Methods for Bond Dissociation Energies of Platinum-Containing Molecules [20]
| Method Category | Specific Method | Mean Average Deviation (MAD) from Experiment (kcal/mol) | Relative Computational Cost |
|---|---|---|---|
| Coupled Cluster (CC) | CCSD(T) | 8.87 | Very High |
| Double Hybrid DFAs | Various (Top 10) | >6.97 | High |
| Hybrid DFAs | TPSSh (For Bond Lengths) | 0.3 pm (for lengths) | Medium |
| Meta-GGA DFAs | TPSSVWN5 | 6.97 | Low-Medium |
| Semiempirical (SE) Methods | Various | Requires Improvement [51] | Low |
| Empirical Force Fields (FF) | Various | Requires Improvement [51] | Very Low |
Table 2: Top-Performing Density Functional Approximations (DFAs) for Quadruple Hydrogen Bonds [56]
| Rank | Density Functional Approximation (DFA) | Category | Key Features |
|---|---|---|---|
| 1 | B97M-V with D3BJ | Berkeley Variant | Empirical dispersion correction |
| 2-9 | Other Berkeley Variants | Berkeley Variants | With/without dispersion corrections |
| 10 | Minnesota 2011 Functionals (e.g., M11) with D3 | Minnesota 2011 | Augmented with dispersion correction |
The data reveals several key insights. First, the performance of Density Functional Approximations (DFAs) is highly variable and application-dependent. For instance, while the hybrid functional TPSSh excelled in predicting bond lengths for platinum-containing molecules, the meta-GGA functional TPSSVWN5 was the closest to experimental bond dissociation energies [20]. Similarly, a recent benchmark on quadruple hydrogen bonds identified eight variants of the Berkeley functionals and two Minnesota 2011 functionals as top performers, with the best being B97M-V using an empirical D3BJ dispersion correction [56]. This highlights that no single DFA is universally superior; the choice must be guided by the specific property of interest and the system being studied.
Second, the results confirm a general Pareto-like relationship: methods with higher theoretical rigor, such as CCSD(T), tend to be more accurate but also vastly more computationally expensive [20]. Conversely, less expensive methods like semiempirical approaches and standard force fields often struggle to capture the full physical complexity of non-covalent interactions, especially for out-of-equilibrium geometries, and thus require significant improvement [51]. The most promising approaches for practical applications on large systems appear to be well-parameterized DFAs that include appropriate treatments of dispersion interactions, offering a favorable compromise on the Pareto frontier for many applications [51] [56].
Experimental Protocols for Method Benchmarking
Protocol 1: Generating the QUID Benchmark Dataset
The QUID framework provides a robust methodology for creating benchmark data for ligand-pocket interactions [51]. The protocol is as follows:
- Monomer Selection: Nine large, flexible, chain-like drug molecules (comprising H, N, C, O, F, P, S, Cl) are selected from the Aquamarine dataset to act as host "pockets." Two small monomers—benzene (C6H6) and imidazole (C3H4N2)—are selected to represent common ligand motifs.
- Dimer Generation: The aromatic ring of the small monomer is aligned with a binding site on the large monomer at a distance of 3.55 ± 0.05 Å, creating an initial dimer structure.
- Geometry Optimization: The dimer structure is optimized at the PBE0+MBD level of theory, resulting in 42 equilibrium dimers classified by the folding of the large monomer ('Linear', 'Semi-Folded', 'Folded').
- Non-Equilibrium Sampling: A subset of 16 equilibrium dimers is selected to generate non-equilibrium conformations. This is done by sampling along the non-covalent bond dissociation pathway using a dimensionless factor q (with values of 0.90, 0.95, 1.00, 1.05, 1.10, 1.25, 1.50, 1.75, 2.00), where q=1.00 is the equilibrium geometry. This yields 128 non-equilibrium structures, modeling snapshots of a binding event.
- Benchmark Energy Calculation: Highly accurate interaction energies (Eint) for these 170 systems are computed using the "platinum standard" protocol, achieving agreement of 0.5 kcal/mol between LNO-CCSD(T) and FN-DMC methods [51].
Protocol 2: High-Throughput DFA Assessment
For large-scale benchmarking of density functionals, a standardized workflow is employed:
- System Preparation: A curated set of molecular systems with reliable experimental or high-level theoretical reference data is assembled (e.g., the QUID set, or sets of molecules with known bond lengths and dissociation energies [20]).
- Computational Settings: A consistent and well-defined computational setup is chosen, including basis sets and integration grids, to ensure a fair comparison between methods.
- High-Throughput Calculation: A wide range of DFAs (often over 100, as in [20] and [56]) are used to compute the target properties (e.g., bond lengths, bond dissociation energies, hydrogen bonding energies).
- Error Analysis: The results from each DFA are compared against the reference data. Statistical measures, such as the Mean Average Deviation (MAD), are calculated to quantitatively rank the performance of the methods [20] [56].
- Dispersion Correction: For DFAs that lack inherent dispersion corrections, the performance with and without empirical dispersion corrections (like D3BJ) is often evaluated, as this can dramatically impact accuracy for non-covalent interactions [56].
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Computational Tools for Electronic Structure Benchmarking
| Tool / Resource | Category | Function in Research |
|---|---|---|
| Coupled Cluster Theory [51] [20] | Ab Initio Wave Function Method | Provides "gold standard" reference energies for benchmarking; highly accurate but computationally prohibitive for large systems. |
| Quantum Monte Carlo (QMC) [51] | Ab Initio Wave Function Method | Provides an alternative high-level reference energy; agreement with CC methods establishes a "platinum standard." |
| Density Functional Approximations (DFAs) [20] [56] | Electronic Structure Method | Workhorse methods offering a balance between cost and accuracy; selection is critical and system-dependent. |
| Empirical Dispersion Corrections (e.g., D3BJ) [56] | Computational Correction | Added to DFT calculations to better describe long-range van der Waals interactions, crucial for non-covalent binding. |
| Benchmark Datasets (e.g., QUID, S66, Splinter) [51] | Curated Data | Collections of molecular systems with high-quality reference data for testing and validating computational methods. |
| Symmetry-Adapted Perturbation Theory (SAPT) [51] | Energy Decomposition Analysis | Decomposes interaction energies into physical components (electrostatics, dispersion, etc.), providing insight into binding mechanisms. |
Visualizing the Pareto Frontier in Method Selection
The following diagram illustrates the conceptual Pareto frontier for selecting an electronic structure method, based on the performance data from the cited benchmarks. The optimal choice for a project lies along this curve, dependent on the specific constraints on accuracy and computational budget.
Navigating the speed-accuracy trade-off is a central challenge in computational chemistry and drug design. By leveraging the concept of the Pareto frontier and relying on rigorous, up-to-date benchmarks such as the QUID dataset, researchers can make informed, rational decisions when selecting electronic structure methods. The data clearly indicates that while high-level ab initio methods remain the benchmark for accuracy, carefully selected Density Functional Approximations—particularly those incorporating robust dispersion corrections—often represent the most efficient and effective choice on the Pareto frontier for practical applications involving bond energies and non-covalent interactions in biologically relevant systems. The ongoing development of new benchmarks and functionals promises to further refine this frontier, pushing the boundaries of what is possible in computational drug discovery.
Method Validation: Benchmarking Against Experimental Data and Real-World Applications
The accurate prediction of bond-dissociation enthalpies (BDEs) is fundamental to advancing research in organic chemistry, drug discovery, and materials science, where understanding bond strength informs the prediction of reaction rates, metabolic sites, and regioselectivity [15]. However, the experimental determination of BDEs through techniques like gas-phase radical kinetics or photoionization mass spectrometry is often impractical for high-throughput applications [15]. While numerous computational benchmarks exist, they frequently focus on narrow chemical domains, creating a need for a compact, diverse benchmark to evaluate modern computational methods efficiently [15]. The ExpBDE54 dataset addresses this need as a slim experimental benchmark comprising 54 small molecules, primarily featuring carbon-hydrogen and carbon-halogen bonds, which are highly relevant to practical problems in organic and medicinal chemistry [15]. This guide provides an objective comparison of computational methods evaluated against ExpBDE54, detailing their performance, experimental protocols, and implementation workflows to help researchers select optimal strategies for rapid BDE prediction.
The ExpBDE54 Benchmark Set
ExpBDE54 is a carefully compiled benchmark set of experimental homolytic bond-dissociation enthalpies for 54 small molecules, designed to cover the most relevant bonding motifs in organic and medicinal chemistry [15]. The dataset is curated from authoritative experimental gas-phase BDE measurements tabulated by Blanksby & Ellison, Yu-ran Luo, and Bordwell et al. [15]. Its "slim" nature, while not extensive enough for training machine-learning models, makes it ideal as an external benchmark for rapid method-development efforts [15]. The set almost exclusively comprises carbon-hydrogen and carbon-halogen bonds, providing a focused yet diverse testbed for assessing computational workflows [15]. The full benchmark, including SMILES strings and corresponding experimental BDE values, is publicly available in the Supporting Information of the primary reference [15] [24].
Performance Comparison of Computational Methods
Various computational approaches were evaluated against the ExpBDE54 benchmark, with their performance quantified by root-mean-square error (RMSE) relative to experimental BDEs. Single-point energies were computed using density-functional theory (DFT), semiempirical methods, and neural network potentials, with linear regression corrections applied to account for enthalpic effects [15]. The following tables summarize the accuracy and efficiency of these methods.
Table 1: Performance of Electronic Structure Methods on ExpBDE54
| Method/Functional | Class | RMSE (kcal·mol⁻¹) | Relative Speed |
|---|---|---|---|
| r2SCAN-D4/def2-TZVPPD | mGGA DFT | 3.6 | 1.0x (baseline) |
| ωB97M-D3BJ/def2-TZVPPD | RSH-mGGA DFT | 3.7 | ~2.0x faster |
| B3LYP-D4/def2-TZVPPD | Hybrid DFT | 4.1 | ~2.0x faster |
| r2SCAN-3c | mGGA DFT | ~4.0* | ~2.5x faster |
| ωB97X-3c | RSH-GGA DFT | ~4.5* | >2.5x faster |
| g-xTB//GFN2-xTB | Semiempirical | 4.7 | ~125x faster |
| eSEN-S (OMol25) | Neural Network Potential | 3.6 | Varies by implementation |
| UMA-M (OMol25) | Neural Network Potential | ~5.0* | Varies by implementation |
Note: Values marked with * are estimates based on textual descriptions in [15].
Table 2: Basis Set Performance in DFT BDE Calculations
| Basis Set | ζ-quality | Effect on RMSE | Computational Cost |
|---|---|---|---|
| def2-TZVPPD | 3 | Minimal (baseline) | Baseline |
| def2-QZVP | 4 | Negligible increase | 1.9x higher |
| vDZP | 2 | ≈1.5 kcal·mol⁻¹ increase | 2x faster |
The Pareto frontier of accuracy versus speed is primarily occupied by methods employing triple-ζ basis sets, with g-xTB//GFN2-xTB and OMol25's eSEN Conserving Small defining the optimal trade-offs between computational cost and prediction accuracy [15]. Notably, suitably corrected semiempirical and machine-learning approaches can achieve accuracy comparable to more expensive DFT methods while offering significant speed advantages [15].
Experimental Protocols and Workflows
Computational Methodology
The standard workflow for calculating BDEs against the ExpBDE54 benchmark involves these key stages [15]:
- Initial Structure Generation: Molecular structures are generated from SMILES strings and optimized with GFN2-xTB to serve as a consistent starting point for all subsequent calculations [15].
- Geometry Optimization: The initial structure is optimized with the target method (DFT, semiempirical, or NNP).
- Bond Cleavage and Fragment Optimization: The target bond is cleaved homolytically, creating two doublet fragments. Fragments with more than one atom are optimized using the same method [15].
- Electronic Energy Calculation: The electronic bond-dissociation energy (eBDE) is calculated as the electronic-energy difference between the molecule and its fragments [15].
- Linear Regression Correction: A linear regression is fit to the calculated eBDEs relative to experimental BDEs to correct for systematic errors, including zero-point energy, enthalpy, and relativistic effects [15].
For methods using different theoretical approaches for geometry optimization and single-point energy calculations (denoted by double-slash notation, e.g., Method_SP//Method_Opt), the optimization phase typically accounts for >90% of the total computational time [15].
Workflow Visualization
The following diagram illustrates the standardized computational workflow for BDE prediction and benchmarking against ExpBDE54:
Research Reagent Solutions
Implementing the ExpBDE54 benchmark requires specific computational tools and resources. The following table details essential "research reagents" for electronic structure calculations and their functions in BDE prediction workflows.
Table 3: Essential Computational Tools for BDE Prediction
| Tool/Resource | Type | Primary Function | Application in BDE Workflows |
|---|---|---|---|
| GFN2-xTB | Semiempirical Method | Rapid geometry optimization | Initial and method-specific structure optimization [15] |
| g-xTB | Semiempirical Method | Single-point energy calculation | Fast eBDE computation with accuracy/speed trade-off [15] |
| r2SCAN-3c | DFT Composite Method | All-electron DFT calculation | Balanced accuracy and speed for medium systems [15] |
| Psi4 | Quantum Chemistry Package | DFT computation platform | Performing DFT calculations with various functionals [15] |
| xtb | Semiempirical Program | GFNn-xTB calculations | Execution of GFN0-xTB, GFN1-xTB, and GFN2-xTB methods [15] |
| geomeTRIC | Optimization Library | Geometry optimization | Optimizing molecular structures across different methods [15] |
| OMol25 NNPs | Neural Network Potentials | Energy prediction | Rapid eBDE computation without explicit physics [45] |
| def2-TZVPPD | Basis Set | Electron wavefunction expansion | High-accuracy DFT calculations approaching basis set limit [15] |
The ExpBDE54 benchmark provides an efficient platform for evaluating computational methods for bond-dissociation enthalpy prediction. Performance comparisons reveal distinct optimal use cases: OMol25's eSEN-S delivers exceptional accuracy for systems where precision is paramount, while g-xTB//GFN2-xTB offers the best speed/accuracy trade-off for high-throughput applications [15]. The r2SCAN-3c//GFN2-xTB workflow represents a robust middle ground, providing DFT-level accuracy with reduced computational cost [15]. For researchers prioritizing rapid screening, semiempirical methods with linear corrections provide viable accuracy with orders-of-magnitude speed improvements [15]. The standardized protocols and reagent solutions detailed in this guide enable consistent implementation across diverse research environments, facilitating advancements in drug discovery, synthetic chemistry, and materials science through reliable bond-strength prediction.
The accurate prediction of molecular properties, such as bond energies and redox potentials, is a cornerstone of computational chemistry, with direct implications for drug development and materials science. The performance of any electronic structure method is ultimately judged by its quantitative accuracy against experimental benchmarks, typically measured through statistical indicators like the Root Mean Square Error (RMSE) and the coefficient of determination (R²). This guide provides an objective, data-driven comparison of the quantitative performance of modern computational methodologies, including neural network potentials (NNPs), density functional theory (DFT), semi-empirical quantum-mechanical (SQM) methods, and machine learning (ML) models. The analysis is framed within the broader thesis of evaluating the accuracy of different electronic structure methods, with a particular focus on their applicability in a research and development setting.
The quantitative performance of various methodologies is summarized in the table below, which compiles key statistical metrics for different chemical properties and molecular sets.
Table 1: Quantitative Performance Metrics Across Computational Methodologies
| Methodology | Property | Molecular Set | RMSE | R² | MAE | Source / Reference |
|---|---|---|---|---|---|---|
| UMA-S (NNP) | Reduction Potential | Organometallic (OMROP, N=120) | 0.375 V | 0.896 | 0.262 V | [45] |
| UMA-S (NNP) | Reduction Potential | Main-Group (OROP, N=192) | 0.596 V | 0.878 | 0.261 V | [45] |
| B97-3c (DFT) | Reduction Potential | Main-Group (OROP) | 0.366 V | 0.943 | 0.260 V | [45] |
| B97-3c (DFT) | Reduction Potential | Organometallic (OMROP) | 0.520 V | 0.800 | 0.414 V | [45] |
| GFN2-xTB (SQM) | Reduction Potential | Main-Group (OROP) | 0.407 V | 0.940 | 0.303 V | [45] |
| GFN2-xTB (SQM) | Reduction Potential | Organometallic (OMROP) | 0.938 V | 0.528 | 0.733 V | [45] |
| XGBoost (ML) | Bond Dissociation Energy | Energetic Materials (N=778) | - | 0.98 | 8.8 kJ/mol | [57] |
| Bond-Corrected G3 | Heat of Formation | Diverse Species | 4.9 kJ/mol | - | - | [58] |
| Image-SR Density Model | Electron Density (Errρ) | QM9 Test Set | - | - | 0.16% | [59] |
Key Performance Insights
- NNPs vs. Traditional Methods: The OMol25-trained Neural Network Potential UMA-S demonstrates competitive, and in some cases superior, accuracy compared to low-cost DFT and SQM methods for predicting reduction potentials. Notably, UMA-S achieved a lower MAE (0.262 V) on organometallic species than B97-3c (0.414 V), indicating a particular strength for complex metal-containing systems [45].
- DFT Reliability: The B97-3c functional provides a robust and accurate baseline for main-group chemistry, as evidenced by its high R² (0.943) and low RMSE (0.366 V) on the OROP set [45].
- SQM Limitations: The GFN2-xTB method shows strong performance for main-group molecules (R² of 0.940) but experiences a significant drop in accuracy for organometallic species (R² of 0.528, RMSE of 0.938 V), highlighting a potential limitation for transition metal applications [45].
- Specialized ML Power: For specific properties like bond dissociation energy (BDE), a well-constructed ML model like XGBoost, using a hybrid feature representation, can achieve exceptional accuracy (R² = 0.98, MAE = 8.8 kJ/mol) [57].
- Empirical Correction Efficacy: The application of bond-based corrections to high-level electronic structure methods like G3 theory can significantly improve accuracy, reducing errors in heats of formation to an RMSE of 4.9 kJ/mol [58].
Detailed Experimental Protocols
A critical aspect of comparing performance data is understanding the methodologies that generated them. This section details the experimental protocols cited in the performance tables.
Benchmarking NNPs on Experimental Reduction Potentials
This protocol evaluates the ability of computational methods to predict experimental reduction potentials for main-group and organometallic species [45].
Diagram 1: Reduction potential benchmarking workflow.
- Data Source: Experimental reduction-potential data was obtained from a curated set of 192 main-group (OROP) and 120 organometallic (OMROP) species, including their non-reduced and reduced geometries, and solvent information [45].
- Geometry Optimization: The non-reduced and reduced structures of each species were optimized using each method (NNP, DFT, or SQM). All NNP geometry optimizations were performed using
geomeTRIC1.0.2 [45]. - Solvent Correction: The optimized structures were then processed through the Extended Conductor-like Polarizable Continuum Solvent Model (CPCM-X) to obtain solvent-corrected electronic energies [45].
- Energy Difference Calculation: The predicted reduction potential (in volts) was calculated as the difference in electronic energy (in electronvolts) between the non-reduced and reduced structures [45].
- Statistical Comparison: The accuracy of each method was quantified by calculating the Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and coefficient of determination (R²) against the experimental values [45].
Machine Learning for Bond Dissociation Energy
This protocol describes the development of a high-accuracy ML model for predicting the bond dissociation energy (BDE) of energetic materials (EMs) [57].
Diagram 2: Machine learning workflow for BDE prediction.
- Dataset Construction: A reliable and representative BDE dataset was built from 778 experimental energetic compounds, with BDE values determined via quantum mechanics calculations [57].
- Feature Representation: A hybrid feature strategy was employed, which couples descriptors of the local target bond with global molecular structure characteristics to sufficiently characterize the BDE [57].
- Data Augmentation: To overcome the limitations of a small dataset, pairwise difference regression was used. This technique augments the data by calculating differences between similar molecules and has the added advantage of reducing systematic errors [57].
- Model Training and Validation: The XGBoost algorithm, a powerful gradient-boosting framework, was used to train the prediction model. The model's performance was evaluated using R² and Mean Absolute Error (MAE) [57].
Bond-Based Correction Method
This protocol outlines a simple empirical technique to improve the accuracy of calculated thermochemical properties like heats of formation [58].
- Base Calculations: Select a base electronic structure method (e.g., G3, B3LYP, TPSS). Calculate the uncorrected heat of formation for a training set of molecules with known experimental values [58].
- Define Bond Types: Identify and define the distinct bond types present in the training set molecules (e.g., C-H, C-C, C=O, N=O) [58].
- Optimize Bond Corrections: For each bond type, optimize a correction factor. The objective is to minimize the difference between the calculated (after corrections) and experimental heats of formation across the entire training set. The underlying principle is that the correction to the calculated bond energy is similar for the same type of bond in different molecules [58].
- Apply to New Molecules: To predict the heat of formation of a new molecule, perform the base electronic structure calculation and then add the optimized corrections for all bonds in the molecule [58].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Resources
| Tool / Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| OMol25 NNPs (eSEN, UMA) | Neural Network Potential | Predicts molecular energies & properties across charge/spin states | Fast, accurate property prediction for main-group & organometallics [45] |
| geomeTRIC | Optimization Library | Python library for geometry optimization | Essential for finding stable molecular structures with NNPs [45] |
| CPCM-X | Solvation Model | Implicit solvation model for energy correction | Accounts for solvent effects in redox potential calculations [45] |
| XGBoost | ML Algorithm | Gradient boosting framework for regression/classification | Building high-accuracy predictive models for specific properties (e.g., BDE) [57] |
| Bond-Based Corrections | Empirical Method | Applies bond-specific corrections to calculated energies | Systematically improving accuracy of ab initio/DFT thermochemical data [58] |
| Image-SR Density Model | Density Prediction | Predicts accurate electron density from atomic superposition | Provides high-quality electron densities for property derivation [59] |
| Psi4 | Quantum Chemistry | Software suite for ab initio quantum chemistry | Performing DFT, coupled-cluster, and other electronic structure calculations [45] |
Accurately predicting the binding affinity between a protein target and a small molecule ligand is a cornerstone of computational drug discovery. It enables researchers to virtually screen millions of compounds, prioritizing the most promising candidates for synthesis and experimental testing, thereby dramatically reducing the time and cost of drug development [60]. The field is undergoing a rapid transformation, moving beyond traditional scoring functions to deep learning models that promise greater accuracy and speed [60] [61]. However, this evolution brings new challenges, including the critical need for robust benchmarking free from data bias and the ability to generalize to novel target classes [61]. This guide objectively compares the performance of current state-of-the-art affinity prediction methods, with a particular focus on how modern graph neural networks (GNNs) address the limitations of earlier approaches. The evaluation is framed within the broader thesis of achieving quantitative accuracy in biomolecular interactions, a pursuit that shares fundamental challenges with the prediction of bond energies in computational chemistry.
The landscape of binding affinity prediction is diverse, encompassing methods from physics-based simulations to data-driven deep learning models. Classical scoring functions, embedded in docking tools like AutoDock Vina and GOLD, rely on force-field, empirical, or knowledge-based calculations. While computationally intensive, they often show limited accuracy in predicting absolute binding affinities [61]. Machine learning-enhanced scoring functions marked a significant shift, using features from protein-ligand complexes to predict affinity with greater speed than energy-based calculations [60]. The current state-of-the-art is dominated by deep learning models, particularly Graph Neural Networks (GNNs) and convolutional networks, which learn directly from the 3D structural data of protein-ligand complexes [61].
A pivotal advancement in these modern GNNs is the move from extracting only global features to learning local pocket interaction features. Earlier models often processed protein and ligand features in separate encoders, which limited their ability to capture the critical, fine-grained interactions within the binding pocket [62]. Newer architectures explicitly model these local 3D interactions, which is crucial for achieving high prediction accuracy as the binding event is primarily determined by the local chemical and structural environment [62].
Comparative Performance Analysis of State-of-the-Art Models
To ensure a fair and realistic comparison of model performance, it is essential to address the issue of train-test data leakage. Recent research has revealed that the standard practice of training on the PDBbind database and testing on the Comparative Assessment of Scoring Functions (CASF) benchmark is flawed by substantial data leakage, leading to an overestimation of model generalization capabilities [61]. Nearly half of the CASF test complexes have highly similar counterparts in the training set, allowing models to "cheat" through memorization. The introduction of PDBbind CleanSplit, a curated dataset that rigorously removes these similarities, provides a more stringent and truthful benchmark [61].
The table below summarizes the key characteristics and performance metrics of leading affinity prediction models when evaluated under these more rigorous conditions.
Table 1: Comparison of Key Protein-Ligand Binding Affinity Prediction Models
| Model Name | Core Methodology | Key Differentiating Feature | Reported Performance (PDBbind CleanSplit) | Generalization Assessment |
|---|---|---|---|---|
| GEMS (Graph neural network for Efficient Molecular Scoring) [61] | Graph Neural Network (GNN) with transfer learning from language models. | Sparse graph modeling of protein-ligand interactions. | State-of-the-art on CASF benchmark; maintains high performance post-CleanSplit. | Robust generalization to strictly independent test sets. |
| PLAGCA [62] | GNN with Graph Cross-Attention mechanism. | Integrates global sequence (FASTA/SMILES) and local 3D pocket graph features. | Outperforms other state-of-the-art computational methods. | Demonstrated superior generalization capability. |
| GenScore [61] | Graph Neural Network (GNN). | Not specified in detail in the provided context. | Benchmark performance drops substantially when trained on CleanSplit. | Performance previously inflated by data leakage. |
| Pafnucy [61] | 3D Convolutional Neural Network (CNN). | Uses 3D voxelized representation of the binding pocket. | Benchmark performance drops substantially when trained on CleanSplit. | Performance previously inflated by data leakage. |
The data clearly demonstrates that models like GEMS and PLAGCA, which are architecturally designed to focus on genuine protein-ligand interactions, maintain their performance on the cleaned benchmark. In contrast, the performance of other previously top-performing models like GenScore and Pafnucy drops markedly, indicating that their high scores were partly driven by exploiting dataset biases [61].
Experimental Protocols for Rigorous Model Evaluation
For researchers seeking to validate binding affinity models or reproduce benchmark results, adhering to rigorous experimental protocols is paramount. The following workflow, formalized by recent studies, is essential for obtaining reliable and generalizable performance metrics [61].
Diagram 1: Rigorous Model Evaluation Workflow. This flowchart outlines the critical steps for creating a bias-free benchmark and fairly assessing model generalization, based on the PDBbind CleanSplit protocol [61].
Detailed Methodological Steps
Dataset Curation via Structure-Based Filtering: The cornerstone of a fair evaluation is a properly split dataset. The PDBbind CleanSplit protocol uses a multimodal clustering algorithm that assesses:
- Protein Similarity: Using TM-scores to quantify 3D protein structure similarity [61].
- Ligand Similarity: Using Tanimoto scores on molecular fingerprints to quantify ligand chemical similarity [61].
- Binding Conformation Similarity: Using pocket-aligned ligand root-mean-square deviation (RMSD) to quantify the similarity of the binding pose [61]. Any training complex that exceeds similarity thresholds (e.g., Tanimoto > 0.9 for ligands) with a test complex is removed to prevent data leakage [61].
Model Training and Hyperparameter Tuning: Models are trained from scratch on the curated training set of PDBbind CleanSplit. It is critical to avoid using any hyperparameters that were optimized on the original, leaked dataset split, as this can reintroduce bias.
Blind Testing and Benchmarking: The final model is evaluated only once on the held-out test set of PDBbind CleanSplit or on established benchmarks like CASF that have been cleared of overlapping complexes. The primary metrics for evaluation are the Root-Mean-Square Error (RMSE) and the Pearson Correlation Coefficient (R) between the predicted and experimental binding affinities [61].
Table 2: Key Resources for Binding Affinity Prediction Research
| Resource Name | Type | Primary Function in Research |
|---|---|---|
| PDBbind Database [61] | Comprehensive Database | Provides a central, curated repository of experimental protein-ligand complex structures and their corresponding binding affinity data for training and testing models. |
| CASF Benchmark [61] | Standardized Benchmark | Offers a widely adopted set of complexes for the comparative assessment of scoring functions, though must now be used with cleaned training sets to ensure fairness. |
| CleanSplit Protocol [61] | Data Curation Tool | A method and filtered dataset split designed to eliminate data leakage and redundancy in PDBbind, enabling a genuine evaluation of model generalization. |
| Graph Neural Network (GNN) Frameworks (e.g., PyTor Geometric, DGL) | Software Tools | Enable the construction and training of GNN models that can natively process the graph-structured data of protein-ligand complexes. |
| Language Models (Protein) | Pre-trained Model | Provide transfer learning of protein representations from vast sequence databases, enriching the feature set for affinity prediction models [61]. |
The field of protein-ligand binding affinity prediction is maturing, with the recognition of data leakage prompting a necessary and healthy shift towards more rigorous evaluation standards. Models like GEMS and PLAGCA represent the vanguard of this shift, demonstrating that architectural innovations focused on local interaction features and robust training on clean data are key to genuine generalization [61] [62]. The pursuit of accuracy in predicting these complex biomolecular interactions mirrors the challenges in quantum chemistry of achieving high accuracy for bond energies, where the choice of electronic structure method and the treatment of electron correlation are critical.
Future progress will likely be driven by several key trends. The integration of protein flexibility and induced fit effects into deep learning models is a major frontier, with approaches like FlexPose and DynamicBind beginning to address the long-standing limitation of treating proteins as rigid bodies [60]. Furthermore, as the focus of drug discovery expands beyond traditional small molecules to include macrocycles, PROTACs, and other large, flexible modalities, affinity prediction models must evolve to handle their increased structural complexity and unique binding modes [60] [63] [64]. Finally, while still nascent, quantum computing holds the potential to revolutionize the underlying electronic structure calculations that form the quantum mechanical basis of molecular interactions, possibly offering exponential speedups for high-accuracy simulations in the future [65].
Understanding and accurately predicting the metabolism of xenobiotics by Cytochrome P450 (CYP450) enzymes represents a fundamental challenge in pharmaceutical research and development. These heme-containing enzymes are the primary catalysts for Phase I metabolism in humans, responsible for the biotransformation of approximately 75% of clinically used drugs [66] [67]. The metabolic fate of a drug candidate, including the potential generation of toxic metabolites, directly impacts its therapeutic efficacy, safety profile, and dosage regimen [66]. Consequently, the ability to precisely predict sites of metabolism (SoMs)—the specific atoms within a molecule where enzymatic transformations occur—has become a critical objective in early-stage drug design. This capability allows medicinal chemists to proactively optimize lead compounds by blocking metabolically labile sites, thereby improving metabolic stability and reducing the risk of costly late-stage failures [66].
The prediction of SoMs is intrinsically linked to understanding the underlying chemical reactivity and bond dissociation energies (BDEs) of potential metabolic sites. CYP450 enzymes primarily facilitate oxidative reactions, such as aliphatic hydroxylation, aromatic epoxidation, and heteroatom dealkylation, which often involve the cleavage of specific chemical bonds [68]. Therefore, the accuracy of SoM prediction is fundamentally dependent on the precision of the electronic structure methods used to compute these thermodynamic properties. This case study examines the performance of various computational chemistry methods for predicting CYP450-mediated metabolism sites, benchmarking their accuracy against experimental data and providing a structured framework for selecting appropriate methodologies based on specific research requirements.
Computational Methodologies for Metabolism Prediction: A Comparative Framework
Computational approaches for predicting CYP450 metabolism have evolved into several distinct paradigms, each with unique strengths and limitations. These can be broadly categorized into three methodological families:
Quantum Chemical (QC) Methods: These approaches use first-principles calculations to model the electronic structure of molecules and predict the thermodynamic and kinetic parameters of metabolic reactions. They range from highly accurate but computationally expensive composite methods to more efficient Density Functional Theory (DFT) functionals [69].
Machine Learning (ML) and Deep Learning (DL) Models: Data-driven models trained on large datasets of known CYP450 substrates and their metabolic profiles. These include traditional QSAR models as well as more advanced graph neural networks (GNNs) that directly learn from molecular structures [66] [68] [67].
Hybrid and Integrated Platforms: Recent efforts have focused on combining multiple approaches into unified frameworks. For instance, DeepMetab integrates GNNs with quantum-informed descriptors and reaction rules to provide end-to-end metabolism prediction [68].
Table 1: Classification of Computational Methods for CYP450 Metabolism Site Prediction
| Method Category | Representative Tools/Methods | Primary Approach | Key Advantages |
|---|---|---|---|
| Quantum Chemical | G4, CBS-QB3, ωB97X-D, MC-PDFT | First-principles electronic structure calculation | High physical fidelity; Mechanistic insight; No training data required |
| Machine Learning | SMARTCyp, FAME, XenoSite | Data-driven pattern recognition from known substrates | High speed for screening; Handles complex molecular representations |
| Integrated Platforms | DeepMetab, BioTransformer 3.0 | Combines multiple approaches (e.g., QC, ML, rules) | Comprehensive prediction (SoM, metabolites, enzyme specificity) |
Benchmarking Quantum Chemical Methods for Bond Energy Calculations
The accurate prediction of bond dissociation energies (BDEs) forms the physical foundation for understanding metabolic reactivity. Recent benchmark studies have systematically evaluated the performance of various quantum chemical methods for predicting BDEs and enthalpies of formation (ΔfH)—key thermodynamic parameters that correlate with metabolic susceptibility [69].
A comprehensive assessment compared four composite methods (CBS-QB3, G3MP2, G3, and G4) and three Density Functional Theory (DFT) functionals (M06-2X, ωB97X-D, and B2PLYP-D3) across various basis sets for calculating C-Cl and C-Br BDEs in halogenated polycyclic aromatic hydrocarbons [69]. The study found that the G4 composite method delivered the best overall performance for thermodynamic property prediction, closely approaching "chemical accuracy" (∼1 kcal/mol) [69]. However, for larger drug-like molecules where G4 becomes computationally prohibitive, the ωB97X-D functional with the 6-311++G(d,p) basis set emerged as the most accurate DFT alternative for BDE prediction, while ωB97X-D/cc-pVTZ performed best for enthalpy of formation calculations [69].
Table 2: Performance Benchmark of Quantum Chemical Methods for Bond Dissociation Energy (BDE) Prediction
| Method | BDE Accuracy (AMUD*) | ΔfH Accuracy (AMUD*) | Computational Cost | Recommended Application |
|---|---|---|---|---|
| G4 | 0.98 kcal/mol | 1.10 kcal/mol | Very High | Small molecules; Final accuracy validation |
| CBS-QB3 | 1.21 kcal/mol | 1.52 kcal/mol | High | Medium-sized molecules; Kinetic studies |
| ωB97X-D/6-311++G(d,p) | 1.15 kcal/mol | 1.89 kcal/mol | Medium | Drug-sized molecules; Routine BDE screening |
| M06-2X/6-311++G(d,p) | 1.42 kcal/mol | 2.34 kcal/mol | Medium | Geometry optimization; Initial screening |
| B2PLYP-D3/cc-pVTZ | 1.87 kcal/mol | 2.56 kcal/mol | Medium-High | Systems with strong dispersion interactions |
| MC-PDFT (MC23) | N/A | N/A | Medium | Systems with strong static correlation [70] |
AMUD: Average Mean Unsigned Deviation from experimental values [69]
For systems exhibiting significant static correlation—such as transition metal complexes, bond-breaking processes, and molecules with near-degenerate electronic states—conventional DFT methods face fundamental limitations [70]. The recently developed multiconfiguration pair-density functional theory (MC-PDFT), particularly the MC23 functional, addresses these challenges by incorporating kinetic energy density and leveraging a multiconfigurational wavefunction, enabling high accuracy for strongly correlated systems at a lower computational cost than advanced wavefunction methods [70].
Experimental Protocols for Method Benchmarking
Workflow for Quantum Chemical Benchmark Calculations
To ensure reproducible and accurate benchmarking of quantum chemical methods for metabolism prediction, the following standardized computational protocol is recommended:
Molecular System Preparation:
- Construct molecular geometries of parent compounds and corresponding radicals using chemical drawing software or computational chemistry packages.
- Perform initial molecular mechanics-based geometry pre-optimization to eliminate steric clashes.
Geometry Optimization and Frequency Analysis:
- Optimize all structures using the M06-2X/6-311++G(d,p) level of theory, which provides an optimal balance between accuracy and computational cost for organic molecules [69].
- Perform analytical harmonic frequency calculations at the same level to confirm stationary points as minima (no imaginary frequencies) and obtain zero-point vibrational energies (ZPVEs).
- Apply a ZPVE scaling factor of 0.97 for M06-2X-calculated frequencies [69].
Single-Point Energy Calculations:
- Compute single-point energies for all optimized structures using multiple methods:
- High-level composite methods (G4, CBS-QB3) for small reference molecules.
- Selected DFT functionals (ωB97X-D, M06-2X, B2PLYP-D3) with various basis sets (6-311++G(d,p), cc-pVTZ, cc-pVQZ).
- For open-shell systems, employ unrestricted calculations with spin contamination checks.
- Compute single-point energies for all optimized structures using multiple methods:
Thermodynamic Property Calculation:
Statistical Validation:
- Compare computed BDEs and ΔfH against experimental values where available.
- Calculate Average Mean Unsigned Deviation (AMUD) and Average Mean Signed Deviation (AMSD) to assess accuracy and systematic biases [69].
The following workflow diagram illustrates the key stages of this benchmarking protocol:
Validation Framework for Site of Metabolism Prediction
To quantitatively evaluate the performance of SoM prediction methods, the following validation metrics and procedures should be implemented:
Dataset Curation:
- Compile a diverse set of drug molecules with experimentally verified SoMs for major CYP450 isoforms (CYP1A2, 2C9, 2C19, 2D6, 3A4) from authoritative databases such as DrugBank and BRENDA [68] [71].
- Ensure dataset includes both substrates and non-substrates to avoid model bias.
- Resolve conflicting annotations through cross-verification across multiple independent sources [71].
Performance Metrics:
- Calculate TOP-1 and TOP-2 accuracy: Percentage of compounds where the experimentally observed SoM is ranked first or within the top two predicted sites [68].
- Compute Matthew's Correlation Coefficient (MCC) to assess binary classification performance (substrate vs. non-substrate) [71].
- Employ cross-validation and external validation on held-out test sets to evaluate generalizability.
Comparative Benchmarking:
Comparative Performance Analysis of Prediction Methods
Quantitative Accuracy Assessment
Recent comprehensive studies have enabled direct comparison of various computational approaches for CYP450 metabolism prediction. The emerging paradigm shows that integrated deep learning frameworks consistently outperform isolated prediction tools across multiple metrics.
Table 3: Comparative Performance of CYP450 Metabolism Prediction Tools
| Tool/Method | Prediction Scope | TOP-1 SoM Accuracy | TOP-2 SoM Accuracy | Substrate Prediction MCC | Key Limitations |
|---|---|---|---|---|---|
| DeepMetab [68] | Substrate, SoM, Metabolites | 85.2% | 100% (18 FDA drugs) | 0.51-0.72 (varies by isoform) | Complex architecture; High compute for training |
| SMARTCyp [66] | SoM | ~65-70% (est. from literature) | N/A | N/A | Limited to 3 CYP isoforms; No substrate profiling |
| FAME [66] | SoM | ~70-75% (est. from literature) | N/A | N/A | Does not differentiate between isoforms |
| CypRules [66] | Substrate/Inhibitor | N/A | N/A | 0.45-0.65 (est. from literature) | No SoM prediction |
| Quantum (ωB97X-D) [69] | BDE/Reactivity | N/A | N/A | N/A | Computationally intensive; No enzyme specificity |
| MetaPredictor [68] | Metabolites | 57.8% | N/A | N/A | Generates meaningless text; Limited accuracy |
The performance advantage of integrated frameworks like DeepMetab stems from their ability to simultaneously model multiple aspects of the metabolism process. By employing a multi-task graph neural network architecture that incorporates both atom-level and bond-level reactivity features, along with quantum-informed descriptors, these systems achieve a more holistic representation of the structural and electronic determinants of metabolic susceptibility [68]. Furthermore, the integration of expert-curated reaction rules ensures mechanistic consistency during metabolite generation, bridging the gap between data-driven pattern recognition and fundamental chemical principles [68].
Successful implementation of CYP450 metabolism prediction requires access to specialized computational tools, datasets, and software resources. The following table catalogs essential "research reagents" for this field:
Table 4: Essential Research Reagents and Computational Resources for CYP450 Metabolism Prediction
| Resource Category | Specific Tools/Databases | Primary Function | Access Information |
|---|---|---|---|
| Curated Datasets | CYP450 Interaction Dataset [71] | Provides substrates/non-substrates for 6 major CYP isoforms | Scientific Data Journal Supplement |
| Quantum Chemistry Software | Gaussian 09, GAMESS, ORCA | Performs DFT and composite method calculations | Commercial and academic licenses |
| Specialized Prediction Tools | SMARTCyp, XenoSite, SOMP | Predicts Sites of Metabolism (SoMs) | Web servers and standalone versions |
| Integrated Platforms | DeepMetab, BioTransformer 3.0 | End-to-end metabolism prediction | Research implementations |
| Chemical Databases | DrugBank, PubChem, ChEMBL | Source of molecular structures and bioactivity data | Publicly accessible web portals |
| CYP450-Specific Databases | Cytochrome P450 Knowledgebase, SuperCYP | CYP-focused interaction data | Specialized databases with web interfaces |
This systematic comparison of computational methods for predicting CYP450 metabolism sites reveals a clear hierarchy of accuracy and applicability across different methodological approaches. High-level composite methods (G4) and advanced DFT functionals (ωB97X-D) provide the most physically accurate foundation for understanding metabolic reactivity through bond dissociation energy calculations, with AMUD values of ~1 kcal/mol achievable for validated systems [69]. However, for comprehensive metabolism prediction encompassing substrate specificity, SoM identification, and metabolite generation, integrated deep learning frameworks like DeepMetab currently deliver superior performance, achieving up to 100% TOP-2 accuracy for clinically relevant drug molecules [68].
The following decision pathway provides a strategic framework for selecting the most appropriate computational method based on specific research objectives and constraints:
For practical drug discovery applications, we recommend a tiered strategy: employ rapid ML-based tools like SMARTCyp or FAME for high-throughput screening of large compound libraries during early lead optimization, then apply more sophisticated integrated platforms like DeepMetab for advanced candidates requiring comprehensive metabolic profiling [66] [68]. Quantum chemical calculations using validated DFT methods (ωB97X-D/6-311++G(d,p)) should be reserved for investigating specific metabolic transformations where mechanistic insight is required or for validating predictions on critical compounds [69]. This multifaceted approach maximizes both efficiency and accuracy while providing complementary insights from fundamentally different computational paradigms.
As the field advances, key developments in multiconfigurational DFT methods (MC-PDFT) for treating strongly correlated systems [70], the expansion of curated interaction datasets [71], and the incorporation of enzyme-specific structural features through molecular dynamics simulations [72] will further enhance predictive accuracy. These innovations promise to deliver increasingly reliable metabolism predictions, ultimately reducing dependence on resource-intensive experimental screening and accelerating the development of safer, more effective pharmaceutical agents.
Conclusion
The accurate prediction of bond energies is no longer a pursuit of a single, universally superior method, but a strategic selection from a diverse toolkit. Foundational principles confirm that the discrepancy between theoretical and experimental values itself provides valuable chemical insight, such as the degree of covalent character in ionic bonds. The methodological landscape is now defined by practical choices: highly accurate but computationally expensive DFT methods like r2SCAN-D4 for final validation, versus remarkably efficient and sufficiently accurate semiempirical (g-xTB//GFN2-xTB) or neural network potential (eSEN) methods for high-throughput virtual screening. Successful application hinges on the troubleshooting and optimization strategies outlined, particularly the use of linear corrections and appropriate basis sets. Validation against modern, focused benchmarks like ExpBDE54 is essential for establishing confidence in any workflow. For biomedical research, these advances translate directly into an enhanced ability to predict metabolic sites and protein-ligand affinities with reduced computational cost, accelerating the pace of rational drug design. Future directions will likely involve the tighter integration of machine-learning-corrected semiempirical methods and the development of bespoke benchmarks for specific biological interactions, pushing the boundaries of predictive chemistry in clinical research.
References
- 1. Bond Energies - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Chemical_Bonding/Fundamentals_of_Chemical_Bonding/Bond_Energies]
- 2. Bond Strengths And Radical Stability [https://www.masterorganicchemistry.com/2013/08/14/bond-strengths-radical-stability/]
- 3. Unified model for the prediction of thermophysical ... [https://www.sciencedirect.com/science/article/abs/pii/S0038109823001916]
- 4. First-Principles Investigation of Size Effects on Cohesive ... [https://www.mdpi.com/2079-4991/13/16/2356]
- 5. New computational chemistry techniques accelerate the ... [https://news.mit.edu/2025/new-computational-chemistry-techniques-accelerate-prediction-molecules-materials-0114]
- 6. High-Pressure Antiferrodistortive tilts, Cohesive energies ... [https://www.sciencedirect.com/science/article/abs/pii/S2352214325001571]
- 7. Sequential bond energies, electrostatic interactions ... [https://www.sciencedirect.com/science/article/abs/pii/S2210271X25002725]
- 8. Bond energy analysis — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/Bond_energy_analysis.html]
- 9. Calculate cohesive energy density using LAMMPS [https://matsci.org/t/calculate-cohesive-energy-density-using-lammps/59927]
- 10. Lattice Enthalpies and Born Haber Cycles [https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_and_Websites_(Inorganic_Chemistry)/Crystal_Lattices/Thermodynamics_of_Lattices/Lattice_Enthalpies_and_Born_Haber_Cycles]
- 11. Lattice Energy: The Born-Haber cycle [https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_and_Websites_(Inorganic_Chemistry)/Crystal_Lattices/Thermodynamics_of_Lattices/Lattice_Energy%3A_The_Born-Haber_cycle]
- 12. Theoretical vs Experimental Lattice Energy [https://chemistryguru.com.sg/theory-vs-expt-le]
- 13. lattice enthalpy (lattice energy) [https://www.chemguide.co.uk/physical/energetics/lattice.html]
- 14. Revision Notes - Born-Haber cycle and lattice enthalpy [https://www.sparkl.me/learn/ib/chemistry-hl/born-haber-cycle-and-lattice-enthalpy/revision-notes/1475]
- 15. ExpBDE54: A Slim Experimental Benchmark for Exploring the ... [https://www.rowansci.com/publications/expbde54]
- 16. A roadmap from the bond strength to the grain-boundary ... [https://www.nature.com/articles/s41467-025-55921-y]
- 17. factors and the importance of adsorbate effects in... Electronic structure [https://www.nature.com/articles/s41524-022-00846-z?error=cookies_not_supported&code=ca1a35dc-32e8-4f9f-ad34-ae91915a1d94]
- 18. Active learning with non-ab initio input features toward efficient... [https://pubs.rsc.org/en/content/articlelanding/2018/sc/c7sc03422a]
- 19. Tuning the d - Band of Nickel Bimetallic Compounds for Glycerol... Center [https://pmc.ncbi.nlm.nih.gov/articles/PMC11821095/]
- 20. bond lengths and bond dissociation energies [https://link.springer.com/article/10.1140/epjd/e2019-90691-1]
- 21. BSE49, a diverse, high-quality benchmark dataset of ... [https://www.nature.com/articles/s41597-021-01088-2]
- 22. The Electronic Structure and Bonding in Some Small Molecules [https://pmc.ncbi.nlm.nih.gov/articles/PMC11902227/]
- 23. Benchmark Experimental Gas-Phase Intermolecular ... [https://pubmed.ncbi.nlm.nih.gov/32070214/]
- 24. ExpBDE54: A Slim Experimental Benchmark for Exploring ... [https://chemrxiv.org/engage/chemrxiv/article-details/6878fc0f728bf9025ef68c49]
- 25. r2SCAN-D4: Dispersion corrected meta-generalized ... [https://arxiv.org/abs/2012.09249]
- 26. and HF-DFT Calculations, and How Does D4 Dispersion ... [https://www.mdpi.com/1420-3049/27/1/141]
- 27. Benefits of Range-Separated Hybrid and Double- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9393870/]
- 28. The vDZP Basis Set Is Effective For Many Density Functionals [https://www.rowansci.com/publications/vdzp]
- 29. Comparison of the Performance of Density Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10146789/]
- 30. Analysis of the Geometric and Electronic Structure of Spin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8908755/]
- 31. Density functional benchmark for quadruple hydrogen bonds [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp00836k]
- 32. Semi-empirical quantum chemistry method [https://en.wikipedia.org/wiki/Semi-empirical_quantum_chemistry_method]
- 33. Comparative analysis of GFN methods in geometry ... [https://arxiv.org/html/2505.09606v1]
- 34. g-xTB - A General-Purpose Tight-Binding Electronic-Structure ... [https://rowansci.com/features/g-xtb]
- 35. Data-Driven Virtual Screening of Conformational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12120983/]
- 36. Comparison of computational chemistry methods for the ... [https://www.nature.com/articles/s41598-020-79153-w]
- 37. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 38. Comparison of Molecular Geometry Optimization Methods ... [https://www.mdpi.com/2227-7390/9/22/2855]
- 39. Which Optimizer Should You Use With NNPs? | Rowan [https://www.rowansci.com/blog/which-optimizer-should-you-use-with-nnps]
- 40. Computing accurate bond dissociation energies of ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389424003832]
- 41. 11.2: Gaussian Basis Sets [https://chem.libretexts.org/Courses/Pacific_Union_College/Quantum_Chemistry/11%3A_Computational_Quantum_Chemistry/11.02%3A_Gaussian_Basis_Sets]
- 42. Basis set — BAND 2025.1 documentation - SCM [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/Basis_Set.html]
- 43. The vDZP Basis Set Is Effective For Many Density Functionals [https://arxiv.org/html/2411.13253v1]
- 44. Benchmarking Quantum Mechanical Levels of Theory for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11331531/]
- 45. Benchmarking OMol25-Trained Models on Experimental ... [https://rowansci.com/publications/omol25-redox]
- 46. Electronic Configuration — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/Electronic_Configuration.html]
- 47. Response of a General Restricted Open-Shell Hartree–Fock ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12557359/]
- 48. Researchers Target Quantum Advantage in Binding ... [https://thequantuminsider.com/2025/06/30/researchers-target-quantum-advantage-in-binding-energy-calculations/]
- 49. What are the types of SCF? [https://mattermodeling.stackexchange.com/questions/1568/what-are-the-types-of-scf]
- 50. Chatbot-assisted quantum chemistry for explicitly solvated ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08677e]
- 51. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 52. Efficiently Estimating Pareto Frontiers with Cyclic Learning ... [https://www.databricks.com/blog/efficiently-estimating-pareto-frontiers]
- 53. The speed-accuracy tradeoff: history, physiology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4052662/]
- 54. Computational analysis of speed-accuracy tradeoff [https://www.nature.com/articles/s41598-022-26120-2]
- 55. Speed-Accuracy Trade-Off - an overview [https://www.sciencedirect.com/topics/psychology/speed-accuracy-trade-off]
- 56. Density functional benchmark for quadruple hydrogen bonds [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00836k]
- 57. Exploring an accurate machine learning model to quickly ... [https://www.sciencedirect.com/science/article/pii/S2589004224006734]
- 58. Bond-based corrections to semi-empirical and ab initio ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261404020603]
- 59. Image super-resolution inspired electron density prediction [https://www.nature.com/articles/s41467-025-60095-8]
- 60. Beyond rigid docking: deep learning approaches for fully ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406700/]
- 61. Resolving data bias improves generalization in binding ... [https://www.nature.com/articles/s42256-025-01124-5]
- 62. Predicting protein-ligand binding affinity with the graph ... [https://pubmed.ncbi.nlm.nih.gov/40139623/]
- 63. The latest trends in drug discovery [https://www.cas.org/resources/cas-insights/2025-drug-discovery-trends]
- 64. Beyond Small Molecules: How Advances in 3D Modelling ... [https://international-biopharma.com/beyond-small-molecules-how-advances-in-3d-modelling-are-opening-new-frontiers-in-macrocyclic-drug-discovery/]
- 65. Quantum Computing Beyond Ground State Electronic ... [https://arxiv.org/html/2509.19709v1]
- 66. Computational methods and tools to predict cytochrome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6590657/]
- 67. Advancing ADMET prediction for major CYP450 isoforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288271/]
- 68. DeepMetab: a comprehensive and mechanistically informed ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04631a]
- 69. Benchmark calculations for bond dissociation energies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9040899/]
- 70. New method improves quantum chemistry simulations [https://pme.uchicago.edu/news/new-method-improves-quantum-chemistry-simulations]
- 71. Curated CYP450 Interaction Dataset: Covering the Majority ... [https://www.nature.com/articles/s41597-025-05753-8]
- 72. Membrane-embedded substrate recognition by cytochrome ... [https://www.sciencedirect.com/science/article/pii/S0021925820410269]