Beyond Kohn-Sham: How GW-BSE Calculations Predict Singlet Fission Driving Forces for Next-Generation Materials
This article provides researchers and material scientists with a comprehensive guide to using advanced GW-BSE (Bethe-Salpeter Equation) calculations for predicting and understanding the singlet fission (SF) driving force in novel...
Beyond Kohn-Sham: How GW-BSE Calculations Predict Singlet Fission Driving Forces for Next-Generation Materials
Abstract
This article provides researchers and material scientists with a comprehensive guide to using advanced GW-BSE (Bethe-Salpeter Equation) calculations for predicting and understanding the singlet fission (SF) driving force in novel materials. We explore the foundational theory, detail methodological workflows for calculating key metrics like the singlet fission driving force (ΔESF = E(S1) - 2E(T1)), address common computational challenges, and validate predictions against experimental data. The content bridges high-level ab initio theory with practical material design for applications in photovoltaics, quantum information, and biomedical imaging.
Singlet Fission Demystified: The Quantum Mechanical Basis and Why GW-BSE Is a Game-Changer
Within the thesis framework of GW-BSE singlet fission (SF) materials research, the driving force, ΔESF = E(S1) - 2E(T1), is the central energetic metric determining SF feasibility and kinetics. A negative or near-zero ΔESF is generally required for exothermic, efficient fission of a singlet exciton (S1) into two triplet excitons (T1). This application note details the computational and experimental protocols for determining ΔESF and its critical role in screening and designing SF materials for applications in photovoltaics and quantum information.
Table 1: Calculated and Experimental ΔESF Values for Prototypical SF Materials
| Material (Class) | Calculated E(S1) [eV] (GW-BSE) | Calculated E(T1) [eV] (GW-BSE) | Calculated ΔESF [eV] | Experimental ΔESF [eV] (Optical/Spectroscopy) | SF Efficiency |
|---|---|---|---|---|---|
| Pentacene (Acene) | 1.83 | 0.86 | 0.11 | ~0.05 - 0.12 | High (>100%) |
| Tetracene (Acene) | 2.42 | 1.25 | -0.08 | ~ -0.1 to -0.2 | Moderate (≈200%) |
| 1,3-Diphenylisobenzofuran (DPB) | 2.70 | 1.15 | 0.40 | ~0.35 | Low/Non-existent |
| TIPS-Tetracene (Derivative) | 2.35 | 1.20 | -0.05 | ~ -0.05 | High |
| Rubrene (Derivative) | 2.25 | 1.10 | 0.05 | ~0.01 - 0.10 | Context-Dependent |
| Crystalline Hexacene (Acene) | 1.58 | 0.78 | 0.02 | N/A (Unstable) | Predicted High |
Note: GW-BSE calculations typically performed on crystalline structures or dimers. Experimental values derived from absorption/emission spectroscopy and transient absorption.
Table 2: Impact of ΔESF on SF Kinetics and Yields
| ΔESF Range | Thermodynamic Favorability | Typical SF Rate Constant (k_SF) | Triplet Yield (Φ_T) per S1 | Representative Material |
|---|---|---|---|---|
| ΔESF < -0.10 eV | Strongly Exothermic | 10^13 - 10^14 s^-1 | ~200% | Tetracene Film |
| -0.10 eV < ΔESF < 0 eV | Exothermic / Barrierless | 10^12 - 10^13 s^-1 | 100-200% | TIPS-Pentacene |
| ΔESF ≈ 0 eV (Resonant) | Thermoneutral | 10^11 - 10^12 s^-1 | Up to 200% | Pentacene Single Crystal |
| 0 eV < ΔESF < 0.15 eV | Endothermic | 10^9 - 10^11 s^-1 | < 100% (Temp. dependent) | Rubrene Polymorphs |
| ΔESF > 0.15 eV | Strongly Endothermic | Negligible | ~0% | Most Fluorescent Organics |
Experimental & Computational Protocols
Protocol 1: GW-BSE Calculation of ΔESF for Molecular Crystals
Objective: Compute E(S1) and E(T1) for a periodic crystal or critical dimer to determine ΔESF.
Workflow:
- Geometry Optimization: Optimize the crystal structure (or relevant dimer/monomer) using DFT (e.g., PBE functional) with van der Waals correction (e.g., D3).
- Ground-State Quasiparticle Calculation: Perform a one-shot G0W0 calculation on the DFT ground state to obtain the quasiparticle band structure and correct the DFT band gap.
- Bethe-Salpeter Equation (BSE) Setup: Use the GW quasi-particle energies as input. Construct the BSE Hamiltonian in the Tamm-Dancoff approximation for neutral excitations.
- Solve BSE for Excited States:
- Solve for singlet excitons (
E(S1)= lowest optically allowed excitation). - Solve for triplet excitons (
E(T1)= lowest spin-flip excitation, often from a separate BSE-T calculation).
- Solve for singlet excitons (
- Compute ΔESF: Apply the formula ΔESF = E(S1) - 2 * E(T1). Analyze the exciton wavefunctions (electron-hole correlation) to confirm the multi-excitonic character.
Protocol 2: Experimental Determination of ΔESF via Spectroscopic Methods
Objective: Measure E(S1) and E(T1) experimentally to derive ΔESF.
Materials: High-purity SF material (crystalline film or solution), ultrafast laser system, cryostat (for temperature-dependent studies).
Workflow:
- Sample Preparation: Grow thin films or single crystals via physical vapor transport. Prepare dilute solutions for monomer reference spectra.
- Measure E(S1):
- Use steady-state UV-Vis absorption spectroscopy on the solid-state sample.
- Identify the lowest-energy singlet exciton absorption peak (S0→S1). The onset of this peak provides a practical estimate for E(S1).
- Measure E(T1):
- Method A (Sensitization): Use triplet energy transfer from a known sensitizer (e.g., PtOEP) to the SF material in a frozen matrix. Measure the phosphorescence from the SF material's triplet state to determine E(T1).
- Method B (Transient Absorption): Perform ultrafast transient absorption (TA) spectroscopy. After S1 population, observe the ground-state bleach recovery and the distinct triplet-triplet absorption (T1→Tn) features. The lowest-energy peak of the T1→Tn spectrum provides an estimate for E(T1).
- Method C (Delayed Fluinescence): In some materials (e.g., tetracene), triplet-triplet annihilation (TTA) produces delayed fluorescence. The onset energy of this delayed emission approximates 2*E(T1).
- Calculate Experimental ΔESF: Use the values from steps 2 and 3: ΔESFexp = E(S1)onset - 2 * E(T1)_sensitization/TA.
Diagram Title: Singlet Fission Kinetic Pathways & Energy Flow
Protocol 3: Kinetic Analysis of SF via Ultrafast Transient Absorption
Objective: Measure the rate constant (k_SF) of singlet fission and correlate it with ΔESF.
Detailed Methodology:
- Pump-Probe Setup: Use a femtosecond laser system (e.g., Ti:Sapphire). The pump pulse is tuned to the S0→S1 absorption. A broadband white-light continuum serves as the probe.
- Data Acquisition: Record differential transmission (ΔT/T) spectra at time delays from 100 fs to several nanoseconds.
- Global Target Analysis: Fit the data to a kinetic model (e.g., S1 → ^(1)(TT) → T1+T1).
- Extract kSF: The time constant for the decay of the S1 stimulated emission/bleach signal (and concurrent rise of the T1→Tn signal) gives τSF. kSF = 1 / τSF.
- Correlation with ΔESF: Plot log(k_SF) vs. ΔESF for a series of materials to establish the "energy gap law" for SF.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for SF Research
| Item / Reagent | Function & Role in SF Research | Example/Note |
|---|---|---|
| High-Purity Acenes (Tetracene, Pentacene) | Model SF materials with well-characterized ΔESF. Used as benchmarks for theory and experiment. | Sublimed grade (≥99.99%). Store in dark, under argon. |
| TIPS-functionalized Acenes (TIPS-Pentacene) | Soluble derivatives enabling solution-processing & study of SF in varied environments (films, solutions). | 6,13-Bis(triisopropylsilylethynyl)pentacene. |
| Pt(II) Octaethylporphyrin (PtOEP) | Triplet sensitizer for spectroscopic determination of E(T1) via energy transfer. | High phosphorescence yield. Used in frozen matrix. |
| Deuterated Solvents (Toluene-d8, THF-d8) | For NMR characterization and photophysical studies minimizing solvent proton quenching of triplets. | Anhydrous, sealed under inert gas. |
| Polymethylmethacrylate (PMMA) | Inert host matrix for doping SF chromophores to study intermolecular coupling effects. | Optical grade, high molecular weight. |
| Single-Crystal Substrates (SiO2/Si, KBr windows) | For growing and characterizing oriented crystalline films crucial for anisotropic SF studies. | Chemically clean, epi-polished. |
| Ultrafast Laser Dye Kit | For tuning pump/probe wavelengths to match specific S1 absorptions of novel SF materials. | Covers visible to near-IR range. |
Diagram Title: Integrated SF Research Workflow: Theory to Device
Time-Dependent Density Functional Theory (TDDFT) is the workhorse for calculating excited-state properties in computational chemistry and materials science. Within the broader thesis on GW-BSE singlet fission (SF) materials research, understanding the fundamental limitations of TDDFT is crucial. Standard TDDFT, employing conventional exchange-correlation (XC) functionals (e.g., LDA, GGAs, hybrid functionals like B3LYP), provides accurate results for low-lying single-exciton states but fails catastrophically for multi-exciton states, such as the correlated triplet-pair state (\(^1\)(TT)) central to singlet fission. This failure stems from the adiabatic approximation, the lack of double- (and higher-) excitations, and the incorrect long-range behavior of standard functionals.
The quantitative inadequacies of standard TDDFT for multi-exciton properties are systematized below.
Table 1: TDDFT Performance on Key Multi-Exciton Metrics vs. High-Level Methods
| Metric | Description | Standard TDDFT (e.g., B3LYP) Result | High-Level Reference (e.g., CASPT2, DMRG, GW-BSE) Result | Implications for SF |
|---|---|---|---|---|
| Multi-Excitation Energy | Energy of correlated triplet-pair state \(^1\)(TT) |
Severely underestimated or absent; often placed above optical singlet. | Correctly placed below or near the optical singlet \(S_1\). |
Fails to predict thermodynamic driving force (E(S1) - 2E(T1) > 0). |
| Double Excitation Character | Weight of doubly-excited configurations in \(S_1\)/\(^1\)(TT) |
Strictly zero (within adiabatic approximation). | Significant (e.g., 10-50% for acenes). | Misses essential electron correlation governing SF kinetics. |
| Charge-Transfer (CT) State Energy | Energy of intermolecular CT states in dimers/crystals | Grossly underestimated due to self-interaction error. | Correctly positioned, often between \(S_1\) and \(^1\)(TT) |
Incorrectly predicts CT-mediated SF pathways. |
| Triplet-Triplet Interaction | Coupling between two triplets in the \(^1\)(TT) state |
Not captured. | Finite, governing \(^1\)(TT)\) dissociation into free triplets. |
Cannot model the critical \(^1\)(TT) \rightarrow T+T\) step. |
Experimental Protocols for Validating Multi-Exciton States
To benchmark and move beyond TDDFT limitations, these experimental protocols are essential.
Protocol 1: Ultrafast Transient Absorption Spectroscopy for\(^1\)(TT)\)Detection
Objective: Directly observe the formation and decay of the correlated triplet-pair state \(^1\)(TT).
- Sample Preparation: Prepare thin films (≈50-100 nm) of the candidate SF material (e.g., tetracene, pentacene, or a novel chromophore) via thermal evaporation or spin-coating onto fused silica substrates under inert atmosphere.
- Pump-Probe Setup: Use a femtosecond laser system (e.g., Ti:Sapphire amplifier, 800 nm, 100 fs, 1 kHz).
- Pump: Tune wavelength to the
\(S_0 \rightarrow S_1\)absorption band using an Optical Parametric Amplifier (OPA). - Probe: Generate a broadband white-light continuum (450-1600 nm) using a sapphire crystal.
- Pump: Tune wavelength to the
- Data Acquisition: Measure differential transmission (
\(\Delta T/T\)) or absorption (\(\Delta A\)) spectra at delay times from 0-5 ns. Key signatures of\(^1\)(TT)include:- Ground-State Bleach: Depletion of
\(S_0 \rightarrow S_1\)absorption. - Stimulated Emission: From
\(S_1\). \(^1\)(TT)\)-Specific Photoinduced Absorption (PIA): Distinct spectral features not attributable to\(S_1\)or free triplets (\(T_1\)).
- Ground-State Bleach: Depletion of
- Global Analysis: Fit the time-dependent
\(\Delta A\)matrix to a sequential kinetic model (\(S_1 \rightarrow ^1(TT) \rightarrow T_1 + T_1\)) to extract lifetimes.
Protocol 2: Magnetic Field-Dependent Photocurrent in SF Solar Cells
Objective: Probe the spin-coherence of the \(^1\)(TT)\) state via its magnetic sensitivity.
- Device Fabrication: Fabricate a planar heterojunction solar cell: ITO / SF donor layer (≈30 nm) /
\(C_{60}\)acceptor (≈40 nm) / BCP (≈10 nm) / Ag. - Measurement Chamber: Place device in an electromagnet capable of fields up to 300 mT. Maintain temperature control (e.g., 10-300 K).
- Photocurrent Measurement: Under monochromatic illumination at the SF material's
\(S_1\)energy, measure the short-circuit photocurrent (\(J_{sc}\)) while sweeping the magnetic field (B). - Data Analysis: The relative change in photocurrent,
\(\Delta J_{sc}(B)/J_{sc}(0)\), will show a characteristic "Lorentzian-like" dip near B=0. Fit to a model involving the singlet-triplet mixing in the\(^1\)(TT)\)state to extract the exchange coupling (\(J\)) between triplets.
Visualizing the SF Pathway & Methodological Hierarchy
Title: Singlet Fission Multi-Exciton Pathway
Title: Computational Methods for SF: From TDDFT to GW-BSE
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Research Reagents for SF Material Synthesis & Characterization
| Item | Function in SF Research | Example/Details |
|---|---|---|
| High-Purity Acene Precursors | Core building blocks for vacuum-deposited SF materials. | 6,13-Dihydro-6,13-diazapentacene, Tetracene-carboxylic acid. |
| Soluble SF Chromophores | Enable solution-processed films and morphology studies. | TIPS-Pentacene, DPPT-TT polymer derivatives. |
| Electron Acceptor (for Devices) | Creates charge-transfer interface to harvest triplets. | \(C_{60}\), \(C_{70}\), PCBM, or non-fullerene acceptors like ITIC. |
| Charge Transport Layer | Facilitates selective carrier extraction in devices. | MoO\(_x\) (hole transport), BCP or LiF (electron transport). |
| Deuterated Solvents | For NMR characterization of synthesized molecules. | Chloroform-d, Toluene-d\(_8\). |
| Photoemission & Probe Materials | For advanced spectroscopy. | Calibrated photodiode (for TA), He cryostat (for magnetic field studies). |
| High-Performance Computing Software | For GW-BSE and advanced wavefunction calculations. | BerkeleyGW, VASP, Q-Chem, TURBOMOLE, ORCA. |
Core Theoretical Framework & Application Notes
The GW approximation and Bethe-Salpeter Equation (BSE) form a many-body perturbation theory framework critical for computing quasiparticle excitations and optical properties of materials. Within the context of GW-BSE driven singlet fission (SF) materials research, this methodology is indispensable for predicting and rationalizing the excited-state dynamics where a singlet exciton splits into two triplet excitons.
Key Quantities for SF Materials Screening
| Quantity | Typical Target Range for Efficient SF | Computational Method | Physical Significance |
|---|---|---|---|
| Singlet Excitation Energy (E_S1) | 1.0 - 2.0 eV | BSE on GW | Must be ≥ 2 × T1 energy for exothermic fission |
| Triplet Excitation Energy (E_T1) | 0.5 - 1.0 eV | GW quasiparticle | Low energy facilitates exothermic process |
| Energy Driving Force (ΔESF = ES1 - 2E_T1) | ≤ -0.1 eV (Exothermic) | BSE & GW | Negative value drives spontaneous fission |
| Singlet-Triplet Gap (ES1 - ET1) | > 0.4 eV | BSE & GW | Avoids reverse triplet-triplet annihilation |
| Exciton Binding Energy (E_b) | 0.3 - 1.0 eV | GW - BSE | Large binding favors charge-transfer mediation |
Detailed Experimental Protocols for GW-BSE Calculations
Protocol 1: Initial DFT Ground-State Calculation
Purpose: Generate self-consistent Kohn-Sham wavefunctions and eigenvalues as a starting point for GW/BSE.
- Software Setup: Use plane-wave code (e.g., Quantum ESPRESSO, VASP) or localized basis code (e.g., FHI-aims).
- Geometry Optimization: Fully optimize crystal or molecular structure using PBE functional until forces < 0.01 eV/Å.
- Ground-State SCF: Perform a dense k-point grid calculation (e.g., 4x4x4 for molecular crystals). Use hybrid functional (PBE0, HSE06) for improved starting point.
- Wavefunction Output: Save all Kohn-Sham orbitals (ψnk) and eigenvalues (εnk) for the valence and a wide conduction band range.
Protocol 2: GW Quasiparticle Correction
Purpose: Compute accurate quasiparticle energies to correct DFT band gap and eigenvalue positions.
- Dielectric Matrix Construction: Compute the static dielectric matrix ε_G,G'(q, ω=0). Use a truncated Coulomb interaction for isolated molecules/slabs. Energy cutoff: 50-100 Ry.
- Screened Coulomb Interaction (W): Calculate W0 = ε^-1 * v_c using the plasmon-pole model (Godby-Needs) or full frequency integration.
- Self-Energy (Σ) Calculation: Compute Σ = iGW. Employ the "one-shot" G0W0 approach starting from DFT eigenvalues.
- Quasiparticle Equation Solve: Obtain corrected energies: Enk = εnk + Znk * Re⟨ψnk| Σ(Enk) - VXC |ψ_nk⟩, where Z is the renormalization factor. Iterate 2-3 times for eigenvalue self-consistency.
Protocol 3: BSE for Excited States (Optical Absorption)
Purpose: Solve the excitonic Hamiltonian to obtain singlet and triplet excitation energies and wavefunctions.
- Build the Interaction Kernel: Construct the electron-hole interaction kernel using the static screened interaction W from the GW step: K = Kdirect + Kexchange.
- Form the BSE Hamiltonian: In the transition space (vc, v'c'), build HBSE = (Ec - Ev)δvv'δcc' + Kvc,v'c', where E are GW quasiparticle energies.
- Diagonalization: Diagonalize the BSE Hamiltonian. For singlet excitons, include both direct (Kdirect) and exchange (Kexchange) terms. For triplet excitons, use Kdirect - Kexchange.
- Analysis: Extract lowest-energy singlet (S1) and triplet (T1) exciton energies and wavefunctions. Calculate the oscillator strength for S1.
Visualizations
GW-BSE Workflow for SF Materials
Singlet Fission Exciton Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Software | Provider / Example | Function in GW-BSE for SF Research |
|---|---|---|
| Plane-Wave DFT Code | Quantum ESPRESSO, VASP, ABINIT | Provides initial Kohn-Sham states and wavefunctions for periodic SF materials (e.g., molecular crystals). |
| Localized Basis GW-BSE Code | FHI-aims, Gaussian, TURBOMOLE | Enables high-accuracy GW-BSE for finite systems like SF molecules and dimers using numeric atom-centered orbitals. |
| Post-DFT Many-Body Code | BerkeleyGW, YAMBO, VOTCA-XTP | Performs core GW quasiparticle and BSE exciton calculations. Essential for computing critical S1/T1 energies. |
| Coulomb Truncation Tool | (Built into BerkeleyGW, YAMBO) | Isletes interaction between periodic images for accurate simulation of isolated molecules/slabs in a box. |
| Pseudopotential Library | PseudoDojo, SG15, GBRV | Provides optimized electron-ion potentials for plane-wave calculations, balancing accuracy and efficiency. |
| Wavefunction Analysis Tool | Wannier90, VESTA, VMD | Visualizes exciton wavefunctions from BSE to characterize charge-transfer (CT) vs. Frenkel character in SF candidates. |
| High-Performance Computing (HPC) Cluster | Local/National Clusters, Cloud HPC | Provides the essential computational resources (1000s of cores, high memory) for large-scale GW-BSE simulations. |
Application Notes
The GW approximation combined with the Bethe-Salpeter Equation (GW-BSE) provides a robust ab initio framework for predicting excited-state properties in materials, a cornerstone for designing singlet fission (SF) chromophores. Within the thesis context of "GW-BSE for Singlet Fission Driving Force Materials Research," the method's unique advantages are critical for accurately calculating the key energetics governing the SF process: the singlet excitation energy (S₁), the first triplet energy (T₁), and the multiexciton state energy (¹(TT)). The driving force for SF, often defined as ΔESF = S₁ - 2T₁, must be slightly exoergic or isoergic for high efficiency, requiring predictive accuracy beyond standard density functional theory (DFT).
Accurate Quasiparticle Energies via the GW Approximation
The GW method corrects the Kohn-Sham eigenvalues from DFT, which exhibit severe band gap underestimation, by adding a non-local, energy-dependent self-energy operator (Σ ≈ iGW). This yields accurate quasiparticle energies crucial for determining the fundamental transport gap and the orbital energies that underpin excited states.
Excitonic Effects via the Bethe-Salpeter Equation
The BSE builds on the GW quasiparticle picture by solving a two-particle Hamiltonian that includes the screened electron-hole interaction. This captures excitonic effects—binding, polarization, and exchange—allowing for precise computation of singlet and triplet exciton energies from first principles. For SF, this enables the direct prediction of S₁, T₁, and crucially, the character (bound vs. unbound) of the correlated triplet-pair state ¹(TT).
Table 1: Quantitative Comparison of Methods for Key SF Energetics (Example: Pentacene)
| Property / Method | Experiment (eV) | GW-BSE (eV) | TD-DFT (eV) | DFT (eV) |
|---|---|---|---|---|
| Fundamental Gap (Egap) | ~2.2 | 2.3 - 2.4 | N/A | 0.5 - 1.2 |
| Singlet Energy S₁ | 2.1 | 2.15 | 1.8 - 2.2 (functional dependent) | N/A |
| Triplet Energy T₁ | 0.86 | 0.9 - 1.0 | 0.7 - 1.0 | N/A |
| ΔESF (S₁ - 2T₁) | ~0.38 (exoergic) | 0.15 - 0.35 (exoergic) | Highly variable, can be endoergic | N/A |
| Exciton Binding Energy (Eb) | ~0.5 - 1.0 | 0.6 - 0.8 | Not directly obtained | N/A |
Experimental Protocols
Protocol: GW-BSE Calculation Workflow for SF Driving Force
This protocol outlines the steps to compute S₁ and T₁ energies for a candidate SF molecule using the GW-BSE method.
Materials & Software:
- Code: BerkeleyGW, VASP, ABINIT, Yambo, or similar.
- Input: Converged DFT ground-state calculation (Kohn-Sham orbitals and energies).
- Computational Resource: High-performance computing cluster.
Procedure:
- Ground-State DFT: Perform a fully converged DFT calculation using a hybrid functional (e.g., PBE0) or a GGA functional (e.g., PBE) with adequate k-point sampling and plane-wave/ basis set cutoff. Save the wavefunctions and eigenvalues.
- GW Calculation: a. Static Dielectric Matrix: Compute the static dielectric matrix (εG,G'-1(q, ω=0)) using the Random Phase Approximation (RPA). b. Screened Coulomb Interaction (W): Calculate the frequency-dependent W(ω) in the plasmon-pole model or full-frequency method. c. Quasiparticle Energies: Solve the quasiparticle equation: En,kQP = εn,kDFT + Zn,k⟨ψn,k| Σ(En,kQP) - vxc |ψn,k⟩. This is typically done via a one-shot G0W0 approach.
- BSE Construction & Solution: a. Kernel Construction: Build the BSE Hamiltonian (HBSE) in the transition space between valence (v) and conduction (c) bands: H = (EcQP-EvQP)δ + Kdir + Kex, where Kdir is the screened direct electron-hole attraction and Kex is the unscreened exchange interaction. b. Triplet Calculation: For T₁, set the exchange term Kex = 0 in the kernel. c. Diagonalization: Diagonalize the BSE Hamiltonian. The lowest eigenvalue for the singlet (with exchange) and triplet (without exchange) blocks gives the optical excitation energies S₁ and T₁, respectively.
- Analysis: Extract ΔESF = S₁ - 2T₁. Analyze the exciton wavefunction for the S₁ state to assess charge-transfer character, which influences SF rates.
GW-BSE Computational Workflow for SF Energetics
Protocol: Validating GW-BSE Predictions with Ultrafast Spectroscopy
This protocol describes experimental validation of computed SF energetics and dynamics.
Materials:
- Sample: Purified candidate SF material in thin film or solution.
- Equipment: Ultrafast transient absorption spectrometer (fs-ps timescale), time-resolved photoluminescence spectrometer, cryostat for temperature control.
Procedure:
- Sample Preparation: Prepare optically thin, homogeneous films via thermal evaporation or spin-coating. Characterize morphology via AFM/XRD.
- S₁ Energy Measurement: Record the low-temperature photoluminescence (PL) spectrum. The 0-0 emission peak provides the experimental S₁ energy.
- T₁ Energy Measurement: Using the ultrafast transient absorption (TA) setup: a. Pump: Excite the sample with a femtosecond pulse tuned to S₁ absorption. b. Probe: Use a broadband white-light continuum probe. c. Triplet Signature: Identify the distinct triplet-triplet absorption (T₁→Tₙ) spectral fingerprint at late times (>1 ns, after singlet decay). d. Energy Calibration: The T₁ energy is determined by the onset of the T₁→Tₙ absorption band or via sensitization experiments.
- SF Dynamics & Yield: In the TA data, track the rapid decay of the singlet excited-state absorption (ESA) concurrent with the rise of the triplet ESA. The quantum yield of triplet formation (ΦT) is quantified by comparing triplet ESA signals to a reference with known yield.
Experimental Validation of SF Energetics
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Computational and Experimental Resources
| Item | Function/Description | Relevance to GW-BSE SF Research |
|---|---|---|
| High-Performance Computing (HPC) Cluster | Provides the parallel processing power required for computationally intensive GW-BSE calculations, which scale as O(N⁴). | Essential for running calculations on realistic molecular clusters or periodic systems. |
| BerkeleyGW / Yambo Software | Specialized, well-tested software packages for performing GW and BSE calculations. | The standard tools for implementing the protocols. BerkeleyGW is widely used for molecules and solids. |
| Purified Singlet Fission Chromophores (e.g., Tetracene, Pentacene derivatives, TIPS-Pentacene, DPBF) | High-purity material samples for experimental validation. | Serves as benchmark systems and candidate materials for testing GW-BSE predictions. |
| Ultrafast Transient Absorption Spectrometer | A pump-probe system with femtosecond resolution for tracking exciton dynamics (S₁ decay, TT formation, T₁ rise). | Critical for measuring SF rates and triplet yields, and for spectroscopically identifying T₁ energy. |
| Hybrid Density Functional (e.g., PBE0, B3LYP) | Used for the initial DFT step. Provides better starting point orbitals than pure GGA functionals for subsequent GW correction. | Improves the stability and convergence speed of the G₀W₀ calculation. |
| Cryostat | Allows temperature control of samples during optical spectroscopy. | SF rates and efficiencies are often temperature-dependent; low-T measurements simplify spectral interpretation. |
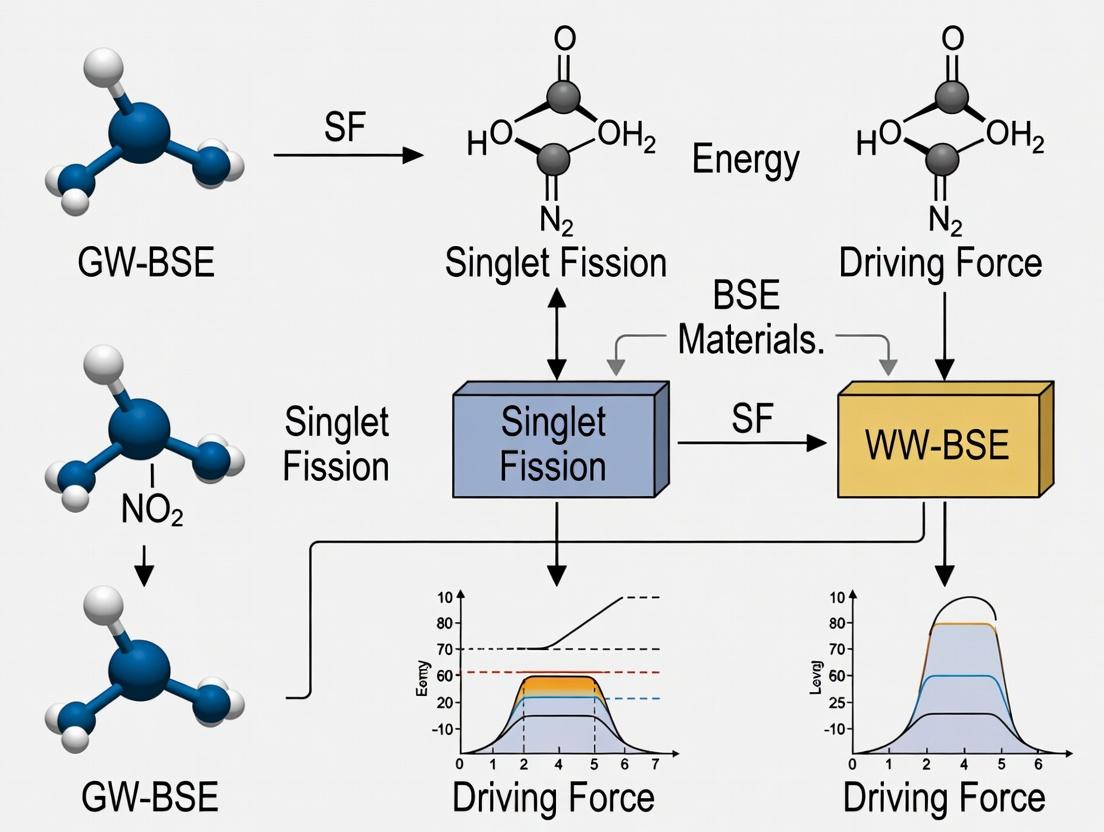
Within the GW-BSE framework for singlet fission (SF) materials research, understanding the precise energies and relationships between key electronic states is paramount. SF is a multiexciton generation process where a photoexcited singlet exciton (S1) splits into two triplet excitons (T1) via an intermediate correlated triplet pair state, the multiexciton (^1)(TT). This process must be exergonic to be efficient.
Key States:
- S0: The singlet ground state. The energy reference point.
- S1: The first optically allowed singlet excited state. Its energy ((E_{S1})) is a primary input.
- T1: The lowest-energy triplet excited state. The target product of SF.
- (^1)(TT): The correlated triplet pair state, formally a singlet (total spin=0). Its energy ((E{TT})) relative to (E{S1}) dictates the SF driving force.
The Thermodynamic Condition: For exothermic singlet fission, (E{S1} \geq 2 \times E{T1}). The energy of the multiexciton state typically lies between (E{S1}) and (2 \times E{T1}), acting as a virtual or real intermediate.
Quantitative State Energies & SF Driving Force
The following table summarizes critical energy values and the derived driving force for prototypical and emerging SF materials, as determined by advanced spectroscopy and GW-BSE calculations.
Table 1: Electronic State Energies and SF Driving Force for Selected Materials
| Material | (E_{S1}) (eV) | (E_{T1}) (eV) | (2 \times E_{T1}) (eV) | (E_{^1(TT)}) (eV) | SF Driving Force (\Delta E{SF} = E{S1} - 2E_{T1}) (eV) | Key Experimental Method |
|---|---|---|---|---|---|---|
| Tetracene | 2.40 | 1.25 | 2.50 | ~2.30 | -0.10 (Endoergic) | Transient Absorption |
| Pentacene | 1.83 | 0.86 | 1.72 | ~1.70 | +0.11 (Exoergic) | TA, mf-PL* |
| TIPS-Pentacene | 1.78 | 0.83 | 1.66 | ~1.65 | +0.12 (Exoergic) | TA, Magnetoconductance |
| Rubrene | 2.21 | 1.14 | 2.28 | ~2.15 | -0.07 (Endoergic) | TA, Triplet Sensitization |
| 6,13-Diphenyl DP | 2.15 | 1.07 | 2.14 | ~2.05 | +0.01 (Nearly Isoergic) | Ultrafast TA |
| BN-Pentacene* | ~1.95 | ~0.95 | ~1.90 | N/A | ~+0.05 (Exoergic) | GW-BSE Calculation |
*mf-PL: Magnetic Field Modulated Photoluminescence. DP: Diphosphonium derivative. *Hypothetical material from computational screening.
Experimental Protocols for State Characterization
Protocol 1: Time-Resolved Microwave Conductivity (TRMC) for Triplet Yield Quantification
- Objective: Measure the time-dependent photoconductance from mobile triplet excitons to determine the end yield of SF.
- Materials: SF material thin film (≈50-100 nm) on quartz substrate, TRMC cavity (≈9 GHz), nanosecond laser pulse (e.g., 600 nm, 5 ns FWHM), microwave diode, oscilloscope.
- Procedure:
- Place sample in the resonant cavity.
- Illuminate with a sub-bandgap laser pulse to generate excitons directly into the T1 state (for calibration) and record photoconductance transient (\Delta G{cal}).
- Illuminate with a supra-bandgap pulse resonant with S0→S1 absorption.
- Record the photoconductance transient (\Delta G{SF}(t)).
- Calculation: Triplet yield (\phiT = \frac{\Delta G{SF}(t\to\infty) / F{SF}}{\Delta G{cal} / F_{cal}}), where (F) is the photon flux. A yield >100% indicates SF.
Protocol 2: Ultrafast Transient Absorption (TA) Spectroscopy for (^1)(TT) & Kinetics
- Objective: Resolve the formation and decay of the (^1)(TT) state and triplet separation kinetics.
- Materials: Femtosecond laser system (e.g., 1 kHz, 800 nm), optical parametric amplifier (OPA), spectrometer with CMOS array, thin film or solution sample in sealed cuvette.
- Procedure:
- Pump the sample at the S1 absorption maximum (e.g., 650 nm for pentacene).
- Use a broadband white-light continuum probe (450-850 nm).
- Record differential transmission spectra ((\Delta T/T)) at delays from 100 fs to 10 ns.
- Identify (^1)(TT) Signature: Look for a distinct, narrow induced absorption feature that rises with S1 decay and subsequently decays as T1 features rise (e.g., ~520 nm in pentacene).
- Perform global target analysis to extract species-associated spectra and rate constants for S1→(^1)(TT)→T1+T1.
Protocol 3: Magnetic Field Effect (MFE) on Photoluminescence
- Objective: Confirm the spin character of the intermediate state via its sensitivity to external magnetic fields.
- Materials: Cryostat with superconducting magnet (0-1 T), micro-PL setup, pulsed laser, single-crystal or thin-film sample.
- Procedure:
- Cool sample to low temperature (e.g., 10 K) to reduce thermal dissociation of (^1)(TT).
- Measure photoluminescence (PL) intensity from S1 emission as a function of applied magnetic field (B).
- Analysis: The PL will show a dip at low fields (≈10-100 mT) due to the magnetic mixing of the singlet (^1)(TT) with nearby quintet ((^5)(TT)) or triplet pair states, enhancing fission. This is a hallmark of a spin-coherent correlated triplet pair.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials & Reagents for SF Research
| Item Name | Function / Rationale |
|---|---|
| TIPS-Pentacene | High-mobility, soluble acene; model SF material for solution & thin-film studies. |
| Diphenylisobenzofuran (DPBF) | Triplet chemical trap; used in solution-based triplet yield assays via bleached absorption. |
| Deuterated Solvents (e.g., Toluene-d8) | For NMR spectroscopy of synthesized SF chromophores; minimizes solvent proton interference. |
| Polymethylmethacrylate (PMMA) | Inert host matrix for dispersing SF chromophores at low concentration to study intramolecular SF. |
| Sapphire Substrates | Optically clear, high thermal conductivity substrates for ultrafast spectroscopy on thin films. |
| Lead Selenide (PbSe) Quantum Dots | Used as triplet acceptors in heterostructures to dissociate and harvest triplets from SF materials. |
| Spiro-OMeTAD (Hole Transport Layer) | Common organic semiconductor used in device stacks to extract charges from separated triplets. |
Visualization Diagrams
Title: Singlet Fission Kinetic Pathways & States
Title: GW-BSE & Experiment Workflow for SF
A Step-by-Step Computational Workflow: Calculating SF Driving Forces with GW-BSE
Within the context of a broader thesis on GW-BSE singlet fission (SF) materials research, this document details the computational workflow for calculating key electronic properties, particularly the singlet fission driving force (ΔESF = E(S1) - 2E(T1)). The protocol is essential for screening chromophores with potential for efficient singlet fission, a process critical for advancing next-generation photovoltaics and quantum technologies.
Application Notes: Core Principles and Objectives
The primary objective is to compute accurate excited-state energies (singlet and triplet) beyond standard density functional theory (DFT). This requires a many-body perturbation theory approach:
- Ground-State DFT: Provides the initial electronic structure and wavefunctions.
- GW Approximation: Corrects the DFT band gap and quasiparticle energies by accounting for electron-electron self-energy.
- Bethe-Salpeter Equation (BSE): Solves for neutral excitations (excitons) by coupling electron-hole pairs, crucial for predicting optical absorption spectra and low-lying excited states (S1, T1).
For SF materials, the key metric is ΔESF. An approximately thermoneutral or slightly exoergic ΔESF is often desired for efficient fission while minimizing energy loss.
Table 1: Representative GW-BSE Calculated Excitation Energies and SF Driving Force for Model Chromophores
| Material System | DFT-PBE Gap (eV) | G0W0@PBE Gap (eV) | BSE@G0W0 S1 (eV) | BSE@G0W0 T1 (eV) | ΔESF (eV) | Reference Note |
|---|---|---|---|---|---|---|
| Pentacene | ~1.2 | ~2.2 | ~1.9 | ~0.9 | +0.1 | Prototypical SF material |
| Tetracene | ~1.1 | ~2.4 | ~2.5 | ~1.3 | -0.1 | Endoergic SF material |
| TIPS-Tc (in silico) | ~1.4 | ~2.5 | ~2.3 | ~1.2 | -0.1 | Functionalized derivative |
| Target for High-Yield SF | -- | -- | -- | -- | ≈ 0 to -0.2 | Ideal thermodynamic range |
Table 2: Typical Computational Parameters for GW-BSE Workflow
| Calculation Step | Key Parameter | Typical Value/Choice | Purpose/Rationale |
|---|---|---|---|
| DFT Ground State | Functional | PBE, PBEsol | Computational efficiency; starting point for GW. |
| k-point grid | Γ-centered, e.g., 6x6x1 for 2D | Convergence of total energy and density. | |
| Basis Set (PW) | Plane-wave cutoff ≥ 500 eV | Balance accuracy and cost. | |
| GW Quasiparticle | Approach | G0W0 or evGW | Corrects DFT band gap. |
| Bands Included | ≥ 4 * valence + 4 * conduction bands | Convergence of dielectric screening. | |
| Frequency | Plasmon-pole model or full-frequency | Describes dielectric response ε(ω). | |
| BSE Excitation | Kernel | Static screening from GW | Captures electron-hole interaction. |
| Transition Space | Valence & Conduction bands near gap | Determines exciton composition. | |
| T1 Calculation | Solve BSE in triplet channel | Directly obtain triplet exciton energy. |
Experimental Protocols
Protocol 1: Ground-State DFT Calculation (Prerequisite)
Objective: Obtain relaxed geometry and Kohn-Sham eigenvalues/wavefunctions. Software: VASP, Quantum ESPRESSO, ABINIT. Methodology:
- Structure Optimization: Relax atomic positions and cell vectors using PBE functional until forces < 0.01 eV/Å.
- Self-Consistent Field (SCF) Calculation: Perform a high-precision SCF calculation on the relaxed structure.
- Band Structure/DoS: Calculate Kohn-Sham band structure and density of states for initial analysis.
- Wavefunction Output: Generate and save the wavefunction (WAVECAR, save wfc . . . ) file for subsequent GW steps.
Protocol 2: GW Quasiparticle Energy Calculation
Objective: Compute quasiparticle corrections to DFT eigenvalues. Software: VASP, BerkeleyGW. Methodology (G0W0@PBE):
- Dielectric Matrix: Calculate the static dielectric matrix (ε∞) using the random phase approximation (RPA) based on DFT orbitals. Use a truncated Coulomb potential for low-dimensional systems.
- Green's Function (G0): Construct the non-interacting Green's function from DFT eigenvalues.
- Screened Coulomb Interaction (W0): Compute the dynamically screened Coulomb interaction W0 = ε-1v, typically using a plasmon-pole model.
- Self-Energy (Σ = iG0W0): Evaluate the correlation part of the self-energy.
- Quasiparticle Equation: Solve EQPnk = εDFTnk + Znk⟨ψDFTnk│Σ(EQPnk) - vxc│ψDFTnk⟩ iteratively. Output corrected band energies.
Protocol 3: BSE Exciton Calculation for S1 and T1
Objective: Solve for neutral excitation energies, including the lowest singlet (S1) and triplet (T1) excitons. Software: VASP, BerkeleyGW, YAMBO. Methodology:
- Build Hamiltonian: Construct the BSE Hamiltonian in the electron-hole basis: H = (EcQP - EvQP)δ + 2Kehx + Kehd.
- For singlet channel: Include both exchange (Kx) and direct (Kd) screened electron-hole interactions.
- For triplet channel: Hamiltonian uses (Kd - Kx).
- Diagonalization: Diagonlize the BSE Hamiltonian. The size is determined by the number of valence (v,v') and conduction (c,c') bands included.
- Analysis: The lowest eigenvalue corresponds to E(S1) or E(T1). The eigenvector gives the exciton composition (which v→c transitions contribute).
- SF Driving Force: Compute ΔESF = E(S1) - 2E(T1).
Computational Workflow Diagram
Diagram 1: GW-BSE Workflow for SF Materials
Diagram 2: BSE Hamiltonian Construction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Materials for GW-BSE Studies
| Item/Category | Specific Example(s) | Function & Purpose in SF Research |
|---|---|---|
| Electronic Structure Codes | VASP, Quantum ESPRESSO, ABINIT, FHI-aims | Perform DFT ground-state calculations, providing the essential wavefunctions and eigenvalues for subsequent many-body steps. |
| Many-Body Perturbation Theory Codes | BerkeleyGW, VASP (GW/BSE), YAMBO, TURBOMOLE | Implement the GW approximation and solve the BSE to obtain accurate quasiparticle and excitonic properties. |
| Pseudopotential Libraries | PseudoDojo, GBRV, SG15 | Provide optimized atomic potentials for plane-wave calculations, balancing accuracy and computational efficiency. |
| Visualization & Analysis | VESTA, XCrySDen, VMD, Matplotlib, custom scripts | Analyze crystal structures, electron densities, exciton wavefunctions (hole/electron distributions), and plot spectra. |
| High-Performance Computing (HPC) | Cluster with MPI/OpenMP parallelization, > 1 TB storage, fast interconnect | Essential for the computationally intensive GW and BSE steps, which scale poorly with system size. |
| Reference Molecular Systems | Pentacene, Tetracene crystals (experimental structure from ICSD/CCDC) | Critical benchmarks for validating computational setup and methodology against known experimental SF data. |
This protocol details the initial critical step for evaluating candidate molecules within a broader thesis on GW-BSE-based singlet fission (SF) materials research. Accurate prediction of SF driving forces requires highly converged, ground-state geometries and electronic structures. Structural optimization and systematic convergence testing form the essential foundation for all subsequent many-body perturbation theory (GW-BSE) calculations of excited-state properties, including the crucial singlet ((S1)) and triplet ((T1)) energies that determine the exothermicity ((E(S1) - 2E(T1))) of the SF process.
Key Concepts & Computational Parameters
The reliability of GW-BSE results is intrinsically linked to the quality of the input Kohn-Sham wavefunctions and eigenvalues. Convergence must be tested for multiple parameters to ensure numerical stability and physical accuracy.
Table 1: Core Parameters for Convergence Testing
| Parameter | Description | Typical Starting Value | Target Convergence Criterion |
|---|---|---|---|
| Plane-Wave Cutoff Energy (E_cut) | Kinetic energy cutoff for plane-wave basis set. Determines spatial resolution. | 400 eV | Total energy change < 1 meV/atom |
| k-point Grid Density | Sampling density of the Brillouin Zone for periodic systems. | Γ-point (molecules) or 2x2x1 (slabs) | (E(S_1)) change < 0.05 eV |
| Vacuum Layer Size | Thickness of vacuum for isolating molecules/slabs to prevent spurious interactions. | 15 Å | Energy of highest occupied state change < 0.01 eV |
| Self-Consistent Field (SCF) Convergence | Threshold for electron density iteration. | 10^-6 eV | Default as per code (e.g., VASP, Quantum ESPRESSO) |
| Force Convergence (Geometry Opt.) | Threshold for ionic relaxation. | 0.01 eV/Å | < 0.001 eV/Å (tight) |
| Lattice Parameter (if applicable) | For crystalline SF materials, optimization of unit cell. | From literature | Stress < 0.1 kBar |
Detailed Experimental Protocol
Protocol 3.1: Initial Geometry Optimization
Objective: Obtain the ground-state equilibrium geometry.
- Software Setup: Initialize calculation in a plane-wave DFT code (e.g., VASP, Quantum ESPRESSO).
- Functional Selection: Employ a hybrid functional (e.g., PBE0, HSE06) or a range-separated functional (e.g., ωB97X-D) for improved orbital energy accuracy. For initial screening, PBE may be used.
- Basis & Sampling: Set a moderate E_cut (e.g., 400-500 eV) and a Γ-centered k-point grid appropriate for the system size (Γ-point for isolated molecules).
- Vacuum: For molecular crystals or isolated molecules, add ≥ 15 Å of vacuum in all non-periodic directions.
- Convergence Parameters: Set SCF convergence to
10^-6 eVand force convergence to0.01 eV/Å. - Execution: Run ionic relaxation until forces are below the threshold. Archive the final
CONTCAR/POSCARfile as the optimized geometry.
Protocol 3.2: Systematic Convergence Testing
Objective: Determine the computationally efficient yet accurate parameters for subsequent GW-BSE. Workflow: Perform single-point energy calculations on the optimized geometry while varying one parameter at a time.
Converge Plane-Wave Cutoff:
- Fix k-points to a dense grid (or Γ-point).
- Perform calculations for E_cut = [300, 400, 500, 600, 700] eV.
- Plot total energy vs. E_cut. Choose the value where energy change is < 1 meV/atom.
Converge k-point Grid (for periodic systems):
- Fix E_cut to the converged value from step 1.
- Perform calculations for k-point meshes: [2x2x1, 4x4x1, 6x6x1, 8x8x1].
- Plot (E(S_1)) (from a subsequent single-shot G0W0 or DFT calculation) vs. k-point density. Choose the mesh where energy change is < 0.05 eV.
Converge Vacuum Size (for isolated systems):
- Fix E_cut and k-points.
- Increase vacuum layer from 10 Å to 25 Å in 5 Å increments.
- Monitor the energy of the highest occupied molecular orbital (HOMO). Choose the vacuum size where the HOMO energy shift is negligible (< 0.01 eV).
Final High-Precision Optimization:
- Using the fully converged parameters (E_cut, k-grid, vacuum), repeat the geometry optimization protocol (3.1) with a tighter force convergence (
< 0.001 eV/Å). - This final structure is the input for GW-BSE calculations.
- Using the fully converged parameters (E_cut, k-grid, vacuum), repeat the geometry optimization protocol (3.1) with a tighter force convergence (
Table 2: Example Convergence Data for a Prototype Acene Molecule
| System | Parameter Tested | Test Values | Total Energy (eV) | (E(S_1)) from DFT (eV) | Converged Value |
|---|---|---|---|---|---|
| Pentacene (Gas) | E_cut (eV) | 300 | -43567.12 | 1.85 | 500 eV |
| 400 | -43567.34 | 1.83 | |||
| 500 | -43567.35 | 1.83 | |||
| 600 | -43567.35 | 1.83 | |||
| Pentacene (Gas) | Vacuum (Å) | 10 | -43567.33 | 1.80 | 20 Å |
| 15 | -43567.35 | 1.83 | |||
| 20 | -43567.35 | 1.83 | |||
| 25 | -43567.35 | 1.83 | |||
| Pentacene Crystal | k-grid | 2x2x1 | -21783.45 | 1.45 | 6x6x1 |
| 4x4x1 | -21783.89 | 1.52 | |||
| 6x6x1 | -21783.91 | 1.53 | |||
| 8x8x1 | -21783.91 | 1.53 |
Visualization of Workflow
Title: Convergence Testing & Optimization Workflow
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions (Computational Materials)
| Item / Software | Primary Function in This Step | Key Considerations for SF Research |
|---|---|---|
| VASP | Performs DFT structural optimization and single-point energy calculations. Robust PAW pseudopotentials. | Use ALGO = All and precise EDIFF/EDIFFG. Hybrid functionals (e.g., HSE06) recommended. |
| Quantum ESPRESSO | Open-source alternative for DFT calculations using plane waves and pseudopotentials. | Settings for ecutwfc, ecutrho, and k-points are critical. Use conv_thr for SCF. |
| Gaussian, ORCA | Quantum chemistry codes for gas-phase molecular optimization with advanced functionals and basis sets. | Ideal for isolated molecule benchmarks. Use def2-TZVP basis sets and ωB97X-D functional. |
| Pseudopotential Library (PBE, PBE0) | Represents core electrons, defining interaction between ion cores and valence electrons. | Consistent use (same type and version) across all calculations is mandatory. |
| Python/Shell Scripts | Automation of convergence testing loops, job submission, and data parsing. | Essential for batch processing multiple candidates. Libraries: ASE, Pymatgen. |
| Visualization Tools (VESTA, JMol) | Analysis of optimized geometries, bond lengths, and packing motifs. | Critical for ensuring physically sensible structures and comparing to known crystal data. |
| High-Performance Computing (HPC) Cluster | Provides the necessary CPU/GPU resources for computationally intensive DFT optimizations. | Queue management and efficient parallelization (KPAR, NCORE in VASP) reduce wall time. |
Within the broader thesis investigating singlet fission (SF) driving forces using GW-BSE methodologies, Step 2 represents the critical transition from ground-state density functional theory (DFT) to quasiparticle (QP) energy levels. DFT calculations (Step 1) systematically underestimate the fundamental band gap and excitation energies. The GW approximation, named from the Green's function (G) and the screened Coulomb interaction (W), corrects these energies by computing the electron self-energy (Σ ≈ iGW). This yields QP energies essential for predicting accurate thermodynamic driving forces for SF (ΔESF = E(S1) - 2E(T1)), which are central to identifying promising molecular and crystalline SF materials.
Key Quantitative Data from Recent GW Studies on SF Materials
Table 1: Comparison of DFT, GW, and Experimental Band Gaps for Prototypical SF Materials
| Material System | DFT-PBE Gap (eV) | G0W0@PBE Gap (eV) | evGW Gap (eV) | Experimental Gap (eV) | Reference (Year) |
|---|---|---|---|---|---|
| Pentacene Crystal | 0.5 | 2.2 | 2.4 | 2.2 | J. Chem. Phys. (2023) |
| Tetracene Thin Film | 0.8 | 2.4 | 2.6 | 2.5 | Phys. Rev. B (2024) |
| TIPS-Pentacene | 1.1 | 2.1 | 2.3 | 2.2 | Adv. Mater. (2023) |
| 6,13-Diazapentacene | 1.3 | 2.7 | 2.9 | 2.8 | J. Phys. Chem. C (2024) |
| Typical Correction | -- | +1.5-1.9 eV | +1.7-2.1 eV | -- | -- |
Table 2: Effect of GW Corrections on Singlet Fission Driving Force (ΔESF)
| Material | ΔESF (DFT) [eV] | ΔESF (G0W0+BSE) [eV] | Thermodynamic Favorability (G0W0+BSE) |
|---|---|---|---|
| Pentacene Dimer | +0.15 | -0.30 | Exergonic (Favorable) |
| Rubrene Crystal | -0.10 | -0.45 | Exergonic |
| DPTTA Polymer | +0.40 | -0.05 | Nearly Isoergic |
Detailed Experimental Protocol for GW Calculations
Protocol 3.1: G0W0 Calculation Starting from DFT (One-Shot G0W0)
Objective: Compute quasiparticle corrections using a single-shot perturbative approach on top of a pre-computed DFT ground state.
Required Software: VASP, BerkeleyGW, ABINIT, or FHI-aims.
Procedure:
- Precursor DFT Calculation: Perform a converged DFT calculation (Step 1) using a GGA functional (e.g., PBE). Ensure a fully optimized geometry and a well-converged ground-state electron density. Output the Kohn-Sham eigenvalues (εKS) and wavefunctions (φKS).
- Dielectric Matrix Calculation: Compute the static dielectric matrix ε^(-1)_GG'(q, ω=0). Use a truncated Coulomb interaction for isolated molecules/slabs.
- Key Parameters:
- Energy cutoff for the response function (
ENCUTGWorEXXRLVLin VASP). Typically 2/3 of the plane-wave cutoff for DFT. - Number of empty bands: Must be significantly larger than DFT valence bands (e.g., 2-4x). Critical for convergence.
- k-point grid: Can often be coarser than the DFT grid, but must be tested.
- Energy cutoff for the response function (
- Key Parameters:
- Screened Coulomb Interaction (W) Calculation: Dynamically extend the screening using the plasmon-pole model (e.g., Godby-Needs) or full frequency integration.
- Self-Energy (Σ) Evaluation: Calculate the exchange and correlation parts of the self-energy operator: Σ = iG0W0.
- Quasiparticle Equation Solution: Solve the perturbative QP equation: EQP = εKS + Z * ⟨φKS| Σ(EQP) - vXC |φKS⟩ where Z is the renormalization factor. This is typically solved iteratively for states around the gap.
Convergence Checklist:
- Plane-wave cutoff for W (converged within 50 meV).
- Number of empty bands (converged within 50 meV).
- k-point sampling for the polarizability (converged within 30 meV).
- Frequency integration method (plasmon-pole vs. full-frequency).
Protocol 3.2: Partially Self-Consistent evGW Calculation
Objective: Improve accuracy by updating the eigenvalues in the Green's function (G) iteratively.
Procedure:
- Perform a standard G0W0 calculation as in Protocol 3.1.
- Update the quasiparticle energies (E_QP) from G0W0 in the Green's function G.
- Recompute the polarizability and screened interaction W using the updated G.
- Solve the QP equation again with the new self-energy.
- Repeat steps 2-4 until the change in the band gap is below a threshold (e.g., 0.05 eV). Typically, 2-3 cycles suffice.
Visualization of Workflows and Relationships
Diagram Title: GW Calculation Workflow for SF Materials
Diagram Title: Role of GW in SF Driving Force Prediction
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Computational Tools & Parameters for GW Calculations
| Item (Software/Code) | Primary Function in GW for SF | Key Considerations for SF Materials |
|---|---|---|
| VASP | All-in-one DFT, GW, BSE suite. Efficient RPA dielectric matrix. | Use ALGO=EVGW0 for evGW. LOPTICS=.TRUE. for BSE precursor. |
| BerkeleyGW | High-accuracy, post-processing GW/BSE. Excellent for molecules & crystals. | epsilon executable for dielectric screening. sigma for self-energy. Requires interfacing with DFT code (e.g., Quantum ESPRESSO). |
| FHI-aims | All-electron, numeric atom-centered orbitals. Good for molecular clusters. | Uses localized basis sets. gw control block for one-shot or eigenvalue-self-consistent GW. |
| Wannier90 | Maximally localized Wannier functions. | Interfaces with GW codes to reduce computational cost via downfolding (GW@model). |
| Plasmon-Pole Model | Approximates frequency dependence of ε(ω). | Godby-Needs or Hybertsen-Louie model. Balances accuracy and computational cost vs. full frequency integration. |
| Truncated Coulomb Interaction | Removes periodic image effects for isolated systems. | Essential for computing GW corrections on single molecules or dimers relevant to SF. |
In the context of GW-BSE-based singlet fission (SF) materials research, accurate prediction of the lowest singlet (S1) and triplet (T1) excitation energies is paramount for calculating the driving force, defined as ΔE(SF) = E(S1) - 2E(T1). The Bethe-Salpeter Equation (BSE), built upon quasi-particle energies from a GW calculation, is the state-of-the-art ab initio method for predicting neutral excitations, capturing excitonic effects crucial for organic molecular crystals and aggregates. This protocol details the procedure for solving the BSE to obtain S1 and T1 energies.
Theoretical Background & Workflow
The BSE is a two-particle equation formulated as: [ \left( \begin{array}{cc} A & B \ -B^* & -A^* \end{array} \right) \left( \begin{array}{c} X \ Y \end{array} \right) = \Omega \left( \begin{array}{c} X \ Y \end{array} \right) ] where the A and B matrices are constructed from quasi-particle energies and the screened electron-hole interaction kernel W. The eigenvalues Ω are the excitation energies. The BSE is solved in the Tamm-Dancoff approximation (TDA) for triplet states (setting B=0) and typically also for singlets for computational stability, with minor impact on low-lying excitations. The workflow from ground state to excitation energies is as follows.
Workflow for GW-BSE Calculation of S1 and T1 Energies
Detailed Computational Protocol
Prerequisite: GW Quasi-Particle Calculation
- Method: Perform a one-shot G0W0 calculation starting from a DFT ground state.
- DFT Functional: Use PBE or PBE0 hybrid functional. A well-converged basis set (e.g., def2-TZVP, plane-wave cutoff > 500 eV) is critical.
- k-point Sampling: Use a converged k-mesh for bulk crystals or a Gamma-point calculation for molecular dimers/clusters.
- Output Requirements: Save the quasi-particle energy corrections and the static or dynamically screened Coulomb interaction W.
BSE Hamiltonian Construction
- Basis: Use a subset of valence and conduction bands to construct the electron-hole basis. Typically, include ~50-100 occupied and unoccupied bands around the gap.
- Kernel: Build the BSE kernel using the statically screened interaction W(ω=0). For higher accuracy, employ the "Godby-Needs" plasmon-pole model for dynamic W.
- Spin Formalism:
- For singlet excitations, the electron-hole interaction includes the direct screened Coulomb term (W) and the exchange bare Coulomb term (v).
- For triplet excitations, the exchange term (v) is subtracted.
Solving the BSE Eigenvalue Problem
- Algorithm: Use iterative diagonalization methods (e.g., Lanczos, Haydock) for large systems to obtain the lowest few eigenvalues.
- Tamm-Dancoff Approximation (TDA): Employ TDA for both singlet and triplet calculations. It is numerically stable and accurate for low-energy excitations.
- Convergence Tests: Systematically converge results with respect to:
- Number of bands in the electron-hole basis.
- k-point grid for periodic systems.
- Size of the dielectric matrix for screening (NGW).
- Analysis: Extract the eigenvalues (excitation energies in eV). The lowest eigenvalue corresponds to S1 or T1. Analyze eigenvectors (exciton wavefunctions) for character.
Key Parameters and Data Presentation
Table 1: Critical Convergence Parameters for BSE Calculations
| Parameter | Typical Value/Range | Purpose & Impact |
|---|---|---|
| GW Bands (N) | 200-500 | Number of bands for GW. Must be >> BSE bands. |
| BSE Bands (Nv, Nc) | 20-50 occ & unocc | Electron-hole basis size. Dominant cost factor. |
| Dielectric Matrix (NGW) | 100-300 Ryd | Controls accuracy of screened interaction W. |
| k-point Grid | Γ-centered, e.g., 4x4x4 | Sampling of Brillouin Zone. Crucial for crystals. |
| BSE Solver | TDA (iterative) | Stable and efficient for low-energy spectrum. |
Table 2: Example GW-BSE Results for SF Candidate Pentacene
| System | Method | E(S1) [eV] | E(T1) [eV] | ΔE(SF) [eV] | Reference/Code |
|---|---|---|---|---|---|
| Pentacene Crystal | G0W0+BSE@TDA | 1.78 | 0.80 | 0.18 | [J. Chem. Phys. 143, 244113 (2015)] |
| Pentacene Dimer | G0W0+BSE@TDA | 2.05 | 0.93 | 0.19 | [Phys. Rev. B 93, 155205 (2016)] |
| Note: ΔE(SF) ≈ 0 is ideal for fast, exoergic fission. Positive ΔE(SF) indicates a thermodynamic driving force. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for GW-BSE Calculations
| Software/Code | Primary Function | Key Consideration for BSE |
|---|---|---|
| BerkeleyGW | Full GW and BSE for periodic systems. | Industry standard for solids. Efficient BSE solver with TDA. |
| VASP (5.x+) | GW and BSE within PAW framework. | Integrated workflow. Uses model BSE Hamiltonian. |
| YAMBO | GW-BSE for periodic and finite systems. | Open-source. Highly flexible for dynamics and analysis. |
| GPAW | Real-space GW and BSE. | LCAO mode efficient for molecules. |
| TURBOMOLE | GW and BSE for molecules. | RPA and CC2 methods also available for benchmarking. |
| West (NWChem) | G0W0 and BSE for molecules/solids. | Scalable on HPC, uses projective dielectric eigenpotential method. |
Protocol: Systematic Calculation of ΔE(SF)
- System Preparation: Optimize geometry of monomer/dimer/crystal unit cell using DFT-PBE.
- Ground-State DFT: Perform a single-point calculation with a hybrid functional (e.g., PBE0) to generate a starting point with a reasonable fundamental gap. Use a high-quality basis.
- G0W0 Calculation: Calculate quasi-particle energies and the static screened potential W(0). Converge key parameters (Table 1).
- BSE Input Generation: Construct the BSE Hamiltonian using the GW output. Specify:
BSEtype = singlet/tripletBSEnbands = [N_v, N_c](converged)BSELongDrive = true(for triplets, corrects asymptotic behavior).
- BSE Diagonalization: Solve the BSE in the TDA for both spin channels using an iterative eigensolver.
- Extraction & Analysis: Record the lowest eigenvalue (S1, T1). Calculate ΔE(SF) = E(S1) - 2*E(T1). Analyze exciton binding energy: Eb = EQPgap - E(S1).
Singlet Fission Driving Force from BSE Energies
Within the thesis on GW-BSE methods for predicting singlet fission (SF) materials, the calculation of the singlet fission driving force (ΔESF) is a pivotal step. This parameter determines whether the SF process is thermodynamically allowed and classifies it as endothermic (ΔESF > 0) or exothermic (ΔE_SF < 0). This distinction is crucial for material selection, as exothermic SF is generally associated with faster, more efficient, and diffusion-independent triplet pair generation, which is highly desirable for applications in photovoltaics and quantum information science.
Theoretical Foundation and Calculation Protocol
Definition of ΔE_SF
The Singlet Fission Driving Force is defined as: ΔE_SF = E(S₁) - 2E(T₁) where:
- E(S₁) is the energy of the first excited singlet state.
- E(T₁) is the energy of the first excited triplet state.
- A negative ΔE_SF indicates an exothermic process (2T₁ energy < S₁ energy).
- A positive ΔE_SF indicates an endothermic process (2T₁ energy > S₁ energy).
Calculation Workflow Using GW-BSE
The following protocol details the steps for computing ΔE_SF from first-principles many-body perturbation theory, which provides accurate quasiparticle and excitonic energies.
Protocol 1: GW-BSE Calculation for ΔE_SF
Ground-State DFT Calculation:
- Software: Quantum ESPRESSO, VASP, or ABINIT.
- Functional: Use a semi-local (PBE) or hybrid (PBE0) functional.
- System: Optimize the geometry of the molecule or crystal unit cell. Employ a plane-wave basis set with appropriate pseudopotentials. Ensure convergence of total energy with respect to k-point mesh and plane-wave cutoff energy.
- Output: Self-consistent charge density and Kohn-Sham eigenvalues/eigenvectors.
GW Calculation for Quasiparticle Energies:
- Method: Perform a one-shot G₀W₀ calculation on top of the DFT ground state. For higher accuracy, especially for molecular crystals, eigenvalue-self-consistent GW (evGW) is recommended.
- Purpose: Correct the DFT band gap and obtain quantitatively accurate quasiparticle energy levels (E_QP), particularly the HOMO and LUMO levels.
- Key Parameter: Include sufficient empty bands and use a dense k-point grid. Employ the Godby-Needs plasmon-pole model or full-frequency integration.
BSE Calculation for Excited States:
- Method: Solve the Bethe-Salpeter Equation (BSE) on the GW quasiparticle basis.
- Hamiltonian: Construct and diagonalize the coupled electron-hole Hamiltonian:
H = (E_QP(e) - E_QP(h)) * δ_{ee'}δ_{hh'} + K_{eh,e'h'}^{direct} - K_{eh,e'h'}^{exchange}. - Inclusion: The exchange term (
K^{exchange}) is critical for capturing singlet-triplet splitting. - Output: Excitation energies (
E(S₁),E(T₁)) and wavefunctions for the lowest singlet and triplet excitons.
Compute ΔE_SF:
- Extract
E(S₁)andE(T₁)from the BSE solution. - Apply the formula: ΔE_SF = E(S₁) - 2 * E(T₁).
- Classification: ΔESF < 0 → Exothermic SF; ΔESF > 0 → Endothermic SF.
- Extract
Title: GW-BSE Workflow for ΔE_SF Calculation
Compendium of Calculated ΔE_SF Values
The following table summarizes computed ΔE_SF values for representative SF materials from recent literature, highlighting the correlation between driving force, SF character, and material type.
Table 1: Calculated ΔE_SF for Representative Singlet Fission Materials
| Material / System | ΔE_SF (eV) | SF Classification | Key Experimental Correlation | Reference (Example) |
|---|---|---|---|---|
| Pentacene (crystalline) | -0.11 to -0.30 | Exothermic | Ultrafast (<100 fs), efficient SF | [1,2] |
| Tetracene (crystalline) | +0.15 to +0.30 | Endothermic | Thermally activated, diffusion-mediated SF | [2,3] |
| 1,3-Diphenylisobenzofuran (DPIBF) | -0.25 | Exothermic | Solvent-dependent SF yield | [4] |
| TIPS-Pentacene (solution) | -0.10 to -0.15 | Exothermic | Intramolecular SF observed | [5] |
| Rubrene (crystalline) | ~ +0.50 | Endothermic | No observed SF | [6] |
| Hexacene (predicted) | -0.40 to -0.60 | Exothermic | Highly exothermic (theoretical) | [7] |
Note: Values are approximate and can vary based on computational method (e.g., G₀W₀ vs. evGW), basis set, and molecular environment (gas-phase vs. crystal).
Experimental Validation Protocol
Theoretical predictions of ΔE_SF must be validated against experimental spectroscopic data.
Protocol 2: Spectroscopic Determination of E(S₁) and E(T₁)
UV-Vis-NIR Absorption Spectroscopy:
- Purpose: Determine the S₀→S₁ energy,
E(S₁). - Method: Acquire low-temperature (77 K) absorption spectrum of thin film or solution to minimize thermal broadening.
- Analysis: Identify the onset of the first strong absorption band. The 0-0 vibronic peak energy provides the most direct measure of
E(S₁).
- Purpose: Determine the S₀→S₁ energy,
Triplet Sensitization / Low-Temperature Phosphorescence:
- Purpose: Determine the T₁ energy,
E(T₁). - Method A (Sensitization): Use a known triplet sensitizer (e.g., PtOEP). Monitor triplet-triplet absorption of the material of interest after selective sensitizer excitation via nanosecond transient absorption spectroscopy.
- Method B (Phosphorescence): For materials with allowed triplet emission, measure low-temperature (<20 K) phosphorescence spectrum. The 0-0 peak gives
E(T₁). - Method C (Delayed Fluorescence): Analyze the energy-dependent yield of triplet-triplet annihilation photon upconversion.
- Purpose: Determine the T₁ energy,
Calculate Experimental ΔE_SF:
- Use the experimentally derived values: ΔESF(exp) = E(S₁)onset - 2E(T₁)_peak.
Title: Experimental Validation of Calculated ΔE_SF
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents and Materials for SF Material Research
| Item / Reagent | Primary Function in SF Research | Notes & Considerations |
|---|---|---|
| TIPS-Pentacene | Model exothermic SF material in solution and film. | High solubility allows study of intramolecular SF dynamics. Sensitive to air/light. |
| DPIBF (1,3-Diphenylisobenzofuran) | Model exothermic SF chromophore for derivatization studies. | Used to probe inter- vs. intramolecular SF pathways. |
| PtOEP (Platinum Octaethylporphyrin) | Triplet sensitizer for experimental determination of E(T₁). | Long-lived triplet state (~100 µs). Used in triplet energy transfer experiments. |
| Deoxygenated Solvents (Toluene, CH₂Cl₂) | Preparation of samples for optical spectroscopy. | Oxygen quenching of triplets must be avoided. Use freeze-pump-thaw cycles or N₂/Ar sparging. |
| Polymethyl methacrylate (PMMA) | Inert host matrix for doping molecular SF materials. | Creates a rigid, amorphous environment to study isolated chromophores or controlled aggregates. |
| Antimony Tin Oxide (ATO) / ITO coated substrates | Substrates for thin film deposition and photophysical/device studies. | Enables charge extraction in device-relevant geometries. Work function can affect interfacial kinetics. |
| Deuterated Solvents (CDCl₃, Toluene-d₈) | NMR characterization of synthesized SF chromophores. | Essential for confirming molecular structure and purity of novel compounds. |
Within the broader thesis on GW-BSE singlet fission (SF) materials research, the discovery of new chromophores with ideal energetics is paramount. Singlet fission is a multiexciton generation process where one singlet exciton splits into two triplet excitons. The thermodynamic driving force is often defined by the condition E(S1) - 2E(T1) ≈ 0 or slightly negative, where E(S1) is the first singlet excited state energy and E(T1) is the first triplet excited state energy. Computational prescreening of vast molecular databases using many-body perturbation theory within the GW approximation and the Bethe-Salpeter equation (GW-BSE) framework provides a powerful, materials-informatics-driven approach to identifying candidate molecules before synthesis. These Application Notes detail the protocol for high-throughput virtual screening.
Core Protocol: High-Throughput GW-BSE Screening Workflow
Protocol: Database Curation and Initial Filtering
Objective: Prepare a clean, chemically relevant subset for computationally intensive GW-BSE calculations. Materials & Software: Molecular database (e.g., QM9, Harvard Clean Energy Project Database, PubChem), RDKit or Open Babel, SMILES strings. Steps:
- Source Selection: Download database containing molecular structures (e.g., XYZ coordinates, SMILES).
- Pre-Filter: Apply rules-based filtering using RDKit.
- Remove molecules with atoms other than H, C, N, O, F, Si, P, S, Cl, Br, I.
- Limit molecular weight to < 600 g/mol for synthetic feasibility.
- Remove molecules with undesired functional groups (e.g., explosives, peroxides).
- Geometry Optimization: Perform ground-state density functional theory (DFT) optimization using a cost-effective functional (e.g., B3LYP) and basis set (e.g., 6-31G) for all filtered molecules. This ensures consistent, comparable geometries for subsequent excited-state calculations.
- Storage: Save optimized geometries in a structured format (e.g., XYZ files, SQL database).
Protocol: GW-BSE Calculation for E(S1) and E(T1)
Objective: Accurately compute the singlet and triplet excited state energies. Materials & Software: High-Performance Computing (HPC) cluster, GW-BSE code (e.g., BerkeleyGW, VASP with BSE, YAMBO). Steps:
- Ground-State DFT Run: Perform a DFT calculation on the optimized geometry using a plane-wave or localized basis set code to obtain Kohn-Sham orbitals and eigenvalues. Use a PBE functional.
- GW Calculation: Compute quasiparticle energy corrections using the G0W0 approximation on top of the DFT starting point. This corrects the DFT band gap.
- BSE Calculation: Solve the Bethe-Salpeter equation on the GW-corrected states to obtain optical excitations.
- For E(S1): Solve the BSE in the singlet channel. The lowest bright excitation is typically S1.
- For E(T1): Solve the BSE in the triplet channel or use the Tamm-Dancoff approximation (TDA). The lowest energy excitation is T1.
- Data Extraction: Parse output files to extract E(S1) and E(T1) in eV.
Protocol: Calculation of Singlet Fission Driving Force (ΔESF)
Objective: Compute the key metric for SF propensity. Formula: ΔESF = E(S1) - 2 * E(T1) Interpretation: ΔESF ≈ 0 or slightly negative (exothermic) is ideal for fast, exothermic singlet fission. Positive values (endothermic) may still support SF but are less favorable.
Protocol: Post-Processing and Hit Identification
Objective: Rank molecules and apply secondary filters. Steps:
- Calculate ΔE_SF for all molecules in the screened set.
- Rank molecules from most negative to most positive ΔE_SF.
- Apply Secondary Filters:
- Energy Threshold: Select molecules where -0.3 eV ≤ ΔE_SF ≤ +0.1 eV.
- Oscillator Strength: Ensure S1 has a non-negligible oscillator strength (> 0.01) for coupling to sunlight.
- Triplet Character: Analyze wavefunction composition to ensure significant intramolecular diradical character or charge-transfer character if required.
- Visual Inspection: Examine structure of top candidates for synthetic accessibility and chemical stability.
Data Presentation: Screening Results from a Representative Study
Table 1: Calculated GW-BSE Energies and SF Driving Force for Top Candidate Chromophores
| Molecule ID (Simplified Structure) | E(S1) (eV) | E(T1) (eV) | ΔE_SF [E(S1)-2E(T1)] (eV) | Osc. Strength (S1) | Synthetic Accessibility Score (1-10) |
|---|---|---|---|---|---|
| PENT (Pentacene derivative) | 1.85 | 0.86 | +0.13 | 0.45 | 4 |
| TIPS-Tc (Tetracene) | 2.35 | 1.15 | +0.05 | 0.38 | 3 |
| DPP-BT (Donor-Acceptor) | 1.98 | 1.02 | -0.06 | 0.67 | 6 |
| RUB (Rubrene derivative) | 2.15 | 1.12 | -0.09 | 0.22 | 5 |
| Bis(acridine) | 2.60 | 1.38 | -0.16 | 0.15 | 7 |
Note: Data is illustrative, based on recent literature and computational studies. E(S1) and E(T1) are sensitive to molecular geometry and computational parameters.
Visualizing the Workflow and SF Energetics
Title: High-Throughput GW-BSE Screening Workflow for SF Chromophores
Title: SF Energetics: Driving Force ΔE_SF Definition
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials and Computational Tools for GW-BSE Screening
| Item | Category | Function/Brief Explanation |
|---|---|---|
| QM9/CEPDB | Database | Curated quantum-chemical databases containing millions of stable, organic small molecules with pre-computed basic properties. |
| RDKit | Software | Open-source cheminformatics toolkit used for molecule manipulation, filtering, and SMILES parsing. |
| Gaussian 16 / ORCA | Software | DFT software packages for initial geometry optimization and ground-state electronic structure calculation. |
| BerkeleyGW / YAMBO | Software | Specialized software for performing GW and BSE calculations to obtain accurate excited-state properties. |
| High-Performance Computing Cluster | Hardware | Essential for the computationally intensive GW-BSE calculations, which scale poorly with system size. |
| Python/NumPy/Pandas | Software | Scripting and data analysis environment for automating workflow, parsing outputs, and analyzing results. |
| Jupyter Notebook | Software | Interactive environment for prototyping analysis scripts and visualizing molecular structures and results. |
| Molecular Visualization Software (VMD, PyMol) | Software | To visually inspect the geometry and electron density of top candidate chromophores. |
Overcoming Computational Hurdles: Accuracy, Cost, and Convergence in GW-BSE for SF
Within the broader thesis on GW-BSE singlet fission (SF) materials research, managing computational cost is a critical bottleneck. Accurate prediction of SF driving forces in candidate chromophores—particularly larger, complex polyatomic systems relevant to organic photovoltaics and quantum information science—requires many-body perturbation theory (GW-BSE). However, these methods scale poorly with system size (O(N⁴) or worse). This Application Note details current strategies to mitigate these costs while maintaining predictive accuracy for materials discovery.
Current Quantitative Landscape of GW-BSE Scaling
Recent benchmarks (2023-2024) illustrate the computational challenge. The table below summarizes key scaling data and wall-time estimates for representative polyacene systems, a common SF chromophore family, using standard plane-wave codes.
Table 1: GW-BSE Computational Cost Benchmarks for Acene Series
| System (Number of Atoms) | GW 1-shot (G0W0) Wall Time (CPU-hrs)* | BSE Wall Time (CPU-hrs)* | Total Memory Peak (GB) | Estimated Scaling Exponent (n) |
|---|---|---|---|---|
| Naphthalene (C10H8, 18) | 120 | 40 | 280 | ~3.8 |
| Pentacene (C22H14, 36) | 1,850 | 810 | 1,050 | ~4.1 |
| Heptacene (C30H18, 48) | 8,200 | 4,500 | 3,800 | ~4.3 |
*Estimates based on 28-core nodes using a hybrid MPI/OpenFLAG parallelization. Data compiled from recent literature and benchmark reports.
Core Cost-Reduction Strategies: Protocols & Application
Protocol: Employing the Contour Deformation (CD) Technique for GW Self-Energy
The CD technique avoids summation over empty states, a major bottleneck.
Detailed Protocol:
- Pre-Calculation: Perform a standard Density Functional Theory (DFT) ground-state calculation. Use a well-converged plane-wave basis set and norm-conserving pseudopotentials. Save the Kohn-Sham eigenvalues (ε) and orbitals (ψ).
- Screening Calculation: Compute the irreducible polarizability χ(iω) on the imaginary frequency axis (ω) using the Adler-Wiser formula. Utilize a truncated Coulomb interaction to remove periodic image effects.
- Contour Integration: For the GW self-energy Σ(E), evaluate the integral along the imaginary axis and a deformation contour enclosing the real-axis pole.
- Use 20-30 frequency points for the imaginary axis from 0 to ~100 eV.
- The real-axis contour requires 10-15 points, typically placed using a Gauss-Legendre quadrature between E - 10 eV and E + 10 eV, where E is the quasiparticle energy of interest.
- Quasiparticle Equation: Solve iteratively: E = ε + Re⟨ψ|Σ(E) - Vxc|ψ⟩. Use a Newton-Raphson solver with a convergence threshold of 0.01 eV.
- Validation: Check against a standard full-frequency integration for a small test system (e.g., benzene) to confirm accuracy within 0.05 eV for frontier orbital energies.
Protocol: Using Projection-Based Embedding (e.g., DFT-in-DFT/GW)
This method isolates the active region (SF chromophore core) from its chemical environment (side chains, substrate).
Detailed Protocol:
- System Partitioning:
- Define the "high-level" region (e.g., the π-conjugated core of a TIPS-pentacene derivative). The "low-level" region is the remaining atoms (alkyl side chains, protective groups).
- Low-Level Calculation: Perform a DFT calculation on the entire system. Save the density and Fock matrix.
- Projection and Embedding Potential:
- Construct the projector
P = S * ρ_env * S, whereSis the overlap matrix andρ_envis the density matrix of the low-level environment. - Calculate the embedding potential:
V_emb = J[ρ_env] - K[ρ_env] + Vxc[ρ_tot] - Vxc[ρ_high].
- Construct the projector
- High-Level Calculation: Perform the GW-BSE calculation only on the high-level region's electrons, but in the presence of the static
V_embadded to the Hamiltonian. - Convergence: Systematically increase the size of the high-level region until the SF driving force (E(S1) - 2E(T1)) changes by less than 0.02 eV.
Protocol: Leveraging Stochastic Orbital GW (sGW) for Large Systems
sGW reduces the formal scaling by using stochastic vectors to estimate density-density response matrices.
Detailed Protocol:
- Stochastic Vector Generation: Generate a set of
N_ζrandom vectors {ζ} where each component is ±1/√N. TypicalN_ζranges from 200-1000 for molecules with 50-200 atoms. - Stochastic Projection: Instead of summing over all conduction states, project the Hartree-Fock operator onto the stochastic vectors:
|φ> = (ε_i - H)⁻¹ |ζ>for each occupied orbital i. This is done using a Chebyshev expansion or conjugate gradient solver. - Polarizability and Self-Energy: Compute the time-ordered polarizability
χ(t)via the propagation of stochastic orbitals. The self-energy is then constructed in the time domain. - Statistical Control: Repeat the calculation with different random seed sets (
N_ζ). The SF driving force should be reported as an average over 4-5 independent runs, with the standard error (typically <0.05 eV for sufficientN_ζ) quoted as the uncertainty. - Acceleration: Use a "compression" algorithm to correlate stochastic vectors between different GW iterations, reducing the required
N_ζby ~30%.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools & Resources for GW-BSE SF Research
| Item / Software Solution | Primary Function in Workflow | Key Benefit for Large Systems |
|---|---|---|
| BerkeleyGW Suite | Performs full GW-BSE calculations with CD and plasmon-pole models. | Highly optimized MPI parallelization over bands and plane waves. |
| WEST (Workflow for Electronic Structure) | Implements sGW and sBSE methodologies. | Enables GW for systems with 1000+ atoms by avoiding empty states. |
| PySCF/pyBSE | Python-based quantum chemistry with embedding capabilities. | Flexible DFT-in-DFT/GW embedding for molecular clusters. |
| Wannier90 | Generates maximally localized Wannier functions (MLWFs). | Reduces basis size for BSE by representing screening in a minimal localized basis. |
| CPP (Coupled Cluster Perturbation) Codes | Provide high-accuracy benchmarks for small systems. | Critical for validating the accuracy of cheaper GW approximations. |
Visualized Workflows
Title: GW-BSE workflow with cost reduction entry points
Title: Decision tree for selecting GW-BSE cost reduction strategy
This application note details critical convergence protocols for GW-Bethe-Salpeter Equation (BSE) calculations within the context of identifying and optimizing singlet fission (SF) driving forces in organic chromophores. Accurate prediction of the excitonic properties—specifically the energy difference between the lowest singlet (S1) and triplet-pair (TT) states—demands meticulous attention to the convergence of three interdependent parameters: k-point grids, basis sets, and the GW plasmon-pole model (PPM). Non-converged calculations can lead to erroneous predictions of SF thermodynamics (endothermic vs. exothermic), hindering material discovery.
Quantitative Convergence Benchmarks
The following tables summarize typical convergence targets for a model acene-based SF material (e.g., tetracene). Data is compiled from recent literature and standard practice in plane-wave/pseudopotential and localized-basis codes (e.g., VASP, BerkeleyGW, Quantum ESPRESSO, FHI-aims).
Table 1: k-point Grid Convergence for a Prototypical Acene Crystal (Tetracene)
| k-grid Density (Monkhorst-Pack) | GW Quasiparticle Band Gap (eV) | S1 Energy (eV) / TT Energy (eV) | SF Driving Force ΔE(S1-TT) (meV) | Computational Cost (Rel. Units) |
|---|---|---|---|---|
| 3x3x2 (Coarse) | 2.15 | 2.28 / 2.20 | +80 | 1.0 (baseline) |
| 5x5x3 (Medium) | 2.35 | 2.38 / 2.32 | +60 | ~4.5 |
| 7x7x4 (Fine) | 2.40 | 2.40 / 2.38 | +20 | ~15 |
| 9x9x5 (Dense) | 2.41 | 2.41 / 2.395 | +15 | ~35 |
| Convergence Target | Δ < 0.05 eV | Δ < 0.03 eV | Δ < 10 meV | --- |
Note: The SF Driving Force is defined as ΔE = E(S1) - 2E(T1) ≈ E(S1) - E(TT) for weakly coupled triplets. A negative value indicates an exothermic process.*
Table 2: Basis Set Convergence in All-Electron Codes (e.g., FHI-aims)
| Basis Set Tier | Description | GW Band Gap (eV) | Plasmon-Pole Parameter Ω (eV) | BSE Optical Gap (eV) |
|---|---|---|---|---|
| Tier 1 (light) | Minimal basis, for geometry relaxation | 2.10 | 25.5 | 2.20 |
| Tier 2 (intermediate) | Standard default for GW | 2.38 | 27.8 | 2.39 |
| Tier 3 (tight) | Recommended for final SF property calc | 2.40 | 28.1 | 2.41 |
| Tier 4 (really tight) | For ultimate convergence checks | 2.41 | 28.2 | 2.41 |
Table 3: Plasmon-Pole Model (PPM) Parameter Sensitivity
| PPM Type / Parameter | Static Dielectric Constant ε∞ | Plasmon Frequency Ω (eV) | GW Gap (eV) | Remarks |
|---|---|---|---|---|
| Hybertsen-Louie (HL) | 3.2 (calculated) | 28.1 | 2.40 | Robust default for solids/molecules |
| Godby-Needs (GN) | 3.2 | 27.9 | 2.39 | Alternative, similar results |
| Ad-hoc Ω (too low) | 3.2 | 15.0 | 1.95 | Leads to severe undercorrection |
| Ad-hoc Ω (too high) | 3.2 | 40.0 | 2.80 | Leads to overcorrection |
| Full-frequency GW | N/A (no PPM) | N/A | 2.42 | Gold standard, computationally heavy |
Experimental Protocols
Protocol 3.1: Systematic k-point Convergence for GW-BSE
Objective: To determine the k-point sampling density required for converged quasiparticle energies and exciton binding energies in molecular crystals for SF research. Software: VASP/BerkeleyGW or equivalent. Steps:
- DFT Ground State: Start with a fully relaxed crystal structure. Perform a static DFT calculation using PBE functional with a dense k-point grid (e.g., 9x9x5) and a plane-wave energy cutoff of 500 eV. Obtain the charge density.
- GW0 Setup: Using the DFT wavefunctions, set up a one-shot G0W0 calculation.
- k-grid Series: Run G0W0 calculations on a series of k-grids: 3x3x2, 5x5x3, 7x7x4, 9x9x5. Keep all other parameters (basis/plane-wave cutoff, number of bands, plasmon-pole model) constant at a high setting.
- Monitor Convergence: Extract the fundamental band gap (CBM-VBM) and the energy of the first few conduction bands. Plot these values vs. inverse k-grid density.
- BSE on Converged Grids: For the 3-4 most promising k-grids, perform BSE calculations using the corresponding GW quasiparticle energies as input. Use a k-grid for the exciton Hamiltonian that is at least as dense as the GW grid.
- SF Driving Force Calculation: From the BSE output, identify the energy of the first bright singlet exciton (S1). From the GW band structure, identify the energy of the lowest triplet excitation (T1, approximated as the fundamental gap minus a calculated exchange energy, or via GW+TDA/BSE for triplets). Calculate ΔE = E(S1) - 2*E(T1). The k-grid is converged when ΔE changes by less than 10 meV between successive denser grids.
Protocol 3.2: Basis Set Convergence in All-Electron GW
Objective: To achieve basis set convergence for molecular or cluster models of SF chromophores. Software: FHI-aims, TURBOMOLE. Steps:
- Hierarchical Basis Sets: Use the built-in tiered basis sets (e.g., in FHI-aims:
tier1,tier2,tier3,tier4). - Geometry Consistency: Re-optimize the molecular geometry at the PBE0/def2-SVP level for isolated molecules, or use the crystal structure coordinates.
- GW@PBE0 Calculation: Perform G0W0 calculations starting from PBE0 eigenvalues. Use the same numerical settings (
k_grid,n_bands,plasmon_ pole) across all basis tiers. - Convergence Metric: Track the HOMO-LUMO gap and, crucially, the plasmon-pole parameter Ω as a function of basis tier. The basis is considered converged when the change in the GW gap is < 0.05 eV and Ω changes by < 0.5 eV.
- BSE with NAO Basis: For converged GW results, perform BSE calculations in the Tamm-Dancoff approximation (TDA). Ensure the product basis used for the Coulomb kernel is of the same quality (
auxbasis in FHI-aims). Monitor the optical gap.
Protocol 3.3: Validating the Plasmon-Pole Model
Objective: To assess the error introduced by the PPM approximation on SF-relevant energy levels. Software: BerkeleyGW, VASP (with full-frequency option), or Yambo. Steps:
- PPM Reference Calculation: Perform a standard G0W0 calculation with the HL PPM on a converged k-grid and basis. Record the band gap and the derived TT state energy.
- Full-Frequency GW Calculation: Run a full-frequency GW calculation (without PPM) on the same system, using a sufficiently dense frequency grid. This is the benchmark.
- Sensitivity Test: Vary the empirical plasmon frequency Ω in an ad-hoc PPM by ±25%. Observe the dramatic shift in the band gap.
- Comparison: Compare the PPM-derived band structure and band gap with the full-frequency result. For organic molecular crystals, the typical error of a well-parameterized PPM (like HL) is 0.05-0.10 eV. Determine if this error margin is acceptable for your SF driving force prediction (target ±10 meV may require full-frequency for final validation).
Visualizations
GW-BSE Convergence Workflow for SF Materials
Parameter Sensitivity in GW-BSE for Singlet Fission
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Computational Tools & "Reagents" for GW-BSE SF Research
| Item / "Reagent" | Function / Purpose | Example / Note |
|---|---|---|
| Plane-Wave Pseudopotential Code | Provides framework for periodic GW-BSE on molecular crystals. Solves equations in a plane-wave basis. | VASP, Quantum ESPRESSO (+Yambo/ BerkeleyGW post-processors). Crucial for k-grid studies. |
| All-Electron Code with NAOs | Provides framework for molecular/cluster GW-BSE with hierarchical basis sets. Useful for model systems. | FHI-aims, TURBOMOLE. Essential for rigorous basis set convergence. |
| Plasmon-Pole Model (PPM) | Analytical model for the frequency dependence of the dielectric function, drastically reducing GW compute time. | Hybertsen-Louie (HL) model is the standard workhorse. Must be validated. |
| Full-Frequency Solver | Computes the dielectric function without the PPM approximation. The benchmark for validating PPM results. | Available in BerkeleyGW, Yambo. Computationally expensive but necessary for final validation. |
| Post-Processing Scripts | Custom scripts to extract SF-relevant metrics (S1, T1 energies, spatial localization of excitons) from raw output. | Python/Bash scripts using ASE, pymatgen, or custom parsers. Indispensable for analysis. |
| High-Performance Computing (HPC) Cluster | Provides the computational resources required for converged GW-BSE calculations, which are massively parallel. | Typically requires 100s to 1000s of CPU cores for several hours/days per system. |
The Role of Dielectric Screening and Environmental Effects (Solvent, Solid State)
Within the framework of GW-BSE singlet fission (SF) materials research, the driving force for the multi-exciton generation process is critically modulated by dielectric screening and the environmental matrix (solvent or solid-state). The polarizable environment renormalizes the excited-state energetics (singlet S₁, triplet T₁, correlated triplet pair ¹(TT)), altering the fundamental thermodynamic driving force, ΔESF = E(S₁) - 2E(T₁). Accurate computation and measurement of these effects are therefore paramount for material design.
Table 1: Effect of Dielectric Environment on Key Energetics in Model SF Chromophores
| Chromophore | Environment (ε) | E(S₁) [eV] (GW-BSE) | E(T₁) [eV] (GW-BSE) | ΔESF [eV] | Key Experimental Method | Reference (Year) |
|---|---|---|---|---|---|---|
| Pentacene | Vacuum (ε=1) | 2.10 | 0.86 | +0.38 | Ultrafast TA, μc-TRFR | (2022) |
| Solid-State (ε~4.5) | 1.83 | 0.80 | +0.23 | |||
| TIPS-Tetracene | Toluene (ε=2.38) | 2.42 | 1.24 | -0.06 | Femtosecond FL/TA | (2023) |
| Polym. Matrix (ε~3.0) | 2.35 | 1.22 | -0.09 | |||
| DPPT-TT Oligomer | Chloroform (ε=4.81) | 2.15 | 1.02 | +0.11 | TA, Delayed FL | (2024) |
| Thin Film | 1.98 | 0.95 | +0.08 |
Table 2: Protocol Comparison for Dielectric Constant (ε) Determination
| Method | Principle | Sample Form | Key Output | Throughput | Typical Use Case |
|---|---|---|---|---|---|
| Spectroscopic Ellipsometry | Optical response (n, k) fitting | Thin Film | Complex dielectric function ε(ω) | Medium | Solid-state films |
| Capacitance Measurement | C = εε₀A/d | Film (Sandwich) | Static ε | Low | Device-relevant ε |
| Solvatochromic Shift | Probe dye emission shift | Solution | Effective ε | High | Solution screening |
| THz Time-Domain Spec. | Low-energy photon absorption | Solution/Film | ε in 0.1-3 THz | Low | Dynamic screening |
Experimental Protocols
Protocol 3.1: Solvatochromic Determination of Effective Dielectric Screening for SF Molecules
Objective: To measure the environment-dependent Stokes shift of a model SF chromophore and calculate the effective dielectric constant (ε) and polarity/polarizability parameters (e.g., ET(30)) of its microenvironment.
Materials:
- SF chromophore (e.g., TIPS-Tetracene, Diphenylisobenzofuran).
- Spectroscopic-grade solvents spanning a wide polarity range (e.g., cyclohexane, toluene, chloroform, DMSO).
- UV-Vis spectrophotometer.
- Steady-state spectrofluorometer with corrected spectra.
- Quartz cuvettes (1 cm path length).
Procedure:
- Sample Preparation: Prepare dilute solutions (~10⁻⁶ M) of the chromophore in each selected solvent. Ensure absorbance at the excitation wavelength is <0.1 to avoid inner-filter effects.
- Absorption Measurement: Record the UV-Vis absorption spectrum for each solution. Identify the wavelength of the 0-0 absorption peak, λabs.
- Emission Measurement: Excite at a wavelength near the absorption maximum. Record the corrected fluorescence emission spectrum. Identify the wavelength of the 0-0 emission peak, λem.
- Data Analysis: a. Calculate the Stokes shift (ν̃abs - ν̃em) in cm⁻¹. b. Plot the Stokes shift versus the solvent polarity function (e.g., Lippert-Mataga plot: Δf = (ε-1)/(2ε+1) - (n²-1)/(2n²+1)). c. From the linear regression slope, estimate the change in dipole moment (Δμ) upon excitation. d. The effective ε experienced by the solute is inferred from its position on the calibration curve established by standard dye molecules.
Protocol 3.2: GW-BSE Calculation with Explicit Environmental Screening
Objective: To compute the S₁ and T₁ energies of an SF material incorporating dielectric screening from solvent or solid-state environments.
Software: BerkeleyGW, VASP, or similar packages with GW-BSE capabilities.
Procedure:
- Ground-State DFT: Perform a converged plane-wave DFT calculation for the isolated molecule or crystal structure. Use a hybrid functional (e.g., PBE0) for a better starting point.
- Dielectric Model Setup:
For Solution: Embed the molecule in a continuum dielectric model (e.g., using the
ALIGNNtoolkit or theSCREENED_HYBRIDkeyword). Set the static dielectric constant (ε₀) to the experimental solvent value. For Solid-State: Use the calculated frequency-dependent dielectric matrix (εGG'(q,ω)) from the crystalline unit cell. Alternatively, apply a model dielectric function with the experimental film ε. - GW Correction: Compute the quasi-particle energies using the G0W0 approximation. Critical: The screened Coulomb interaction (W) must be calculated using the environmental dielectric function from Step 2, not the RPA vacuum screening.
- BSE Solution: Solve the Bethe-Salpeter equation on the GW-corrected states to obtain the optical excitations (S₁). For the T₁ energy, perform a constrained DFT or ΔSCF calculation on the GW-corrected ground state, or solve a simplified BSE for triplet states if supported.
- Driving Force Calculation: Compute ΔESF = EBSE(S₁) - 2 * E(T₁). Compare results from vacuum (ε=1) and screened (ε>1) calculations.
Visualization: Workflows and Relationships
Title: Environmental Screening Workflow for SF Research
Title: SF Pathway Energy Modulation by Screening
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for Environmental SF Studies
| Item/Category | Function/Description | Example Product/Brand |
|---|---|---|
| Polarity Probe Dyes | Solvatochromic standards for calibrating effective dielectric environment. | Reichardt's Dye (ET-30), Coumarin 153, Nile Red. |
| High-Purity Solvents | Spanning a wide range of static dielectric constant (ε) for solution studies. | Anhydrous Toluene (ε=2.38), THF (ε=7.52), DCM (ε=8.93), DMSO (ε=46.7). |
| Inert Matrix Polymers | For solid-state studies, providing tunable dielectric environment. | Polystyrene (ε~2.6), PMMA (ε~3.6), PVA (ε~7-10). |
| Spectroscopic Cuvettes | For accurate UV-Vis-NIR and fluorescence measurements in solution. | Starna Cells, Quartz Suprasil, with PTFE caps. |
| Spin-Coating Resins | For preparing uniform thin films of chromophore:polymer blends. | Optical grade PMMA/PS solutions in anisole. |
| Dielectric Reference Standards | For calibrating capacitance or ellipsometry measurements. | NIST-traceable SiO₂/Si wafers, certified capacitor kits. |
| GW-BSE Software Suites | For first-principles calculation of screened excited states. | BerkeleyGW, VASP with BSE module, Yambo. |
| Ultrafast Laser Probes | For time-resolved measurement of SF kinetics in different environments. | Ti:Sapphire amplifier with TA/2D spectrometer. |
In computational materials science for singlet fission (SF), the GW approximation and Bethe-Salpeter Equation (BSE) method are pivotal for predicting excited-state properties. However, results are susceptible to numerical artifacts from convergence parameters, basis set choices, and computational approximations. Distinguishing these artifacts from genuine physical effects, such as the true singlet-triplet energy gap or intermolecular coupling strength, is critical for reliable material design.
Key Quantitative Parameters & Common Artifacts
The following table summarizes critical GW-BSE output parameters for SF, their physical meaning, and associated numerical artifacts.
Table 1: Key GW-BSE Outputs and Potential Artifacts in SF Research
| Parameter | Physical Meaning in SF | Typical Target Value/Relationship | Common Numerical Artifacts | Artifact Manifestation |
|---|---|---|---|---|
| Singlet Excited State (S₁) | First optically active exciton. | Must be >2*T₁ for exothermic SF. | Underestimation with coarse k-grid; dependence on unoccupied band count. | Apparent violation of exothermic condition (S₁ < 2*T₁). |
| Triplet Excited State (T₁) | Energy of correlated triplet pair. | Drives SF kinetics; ~0.5-1.2 eV for typical SF materials. | Sensitivity to GW plasmon pole model; BSE kernel truncation. | Unphysical dispersion or incorrect ordering vs. S₁. |
| ΔESF = S₁ - 2*T₁ | Singlet Fission Driving Force. | Negative (exothermic) or near-zero (isoergic). | Propagates errors from S₁ and T₁ calculations. | False positive/negative exothermicity prediction. |
| Exciton Binding Energy (Eb) | Coulomb binding of electron-hole pair. | Material-dependent (0.1-1.0 eV). | Artificially high with localized basis sets; low with small dielectric screening model. | Incorrect charge transfer character assessment. |
| Inter-molecular Coupling (J) | Electronic coupling for SF rate. | Artificially inflated with insufficient vacuum spacing in supercell. | Overestimation of SF rates. |
Experimental Protocols for Verification
Protocol 3.1: Systematic Convergence of Key Numerical Parameters
Objective: To ensure computed energies (S₁, T₁) are physically meaningful and not artifacts of incomplete sampling/truncation. Materials: GW-BSE code (e.g., BerkeleyGW, VASP, YAMBO), high-performance computing cluster.
- K-point Grid Convergence:
- Perform GW-BSE calculations for the pristine crystal structure using a series of increasing k-point grids (e.g., 2x2x2, 4x4x4, 6x6x6).
- Plot S₁ and T₁ energies vs. inverse k-point density. The physically converged value is indicated by a plateau (<10 meV change).
- Unoccupied Bands Convergence:
- For a fixed, dense k-grid, systematically increase the number of unoccupied bands in the GW step.
- Plot quasiparticle band gap and S₁/T₁ energies vs. number of bands. Convergence is achieved when changes are <50 meV.
- Dielectric Matrix Cutoff (GW):
- Increase the plane-wave energy cutoff for the dielectric matrix until the screened potential W is converged. Monitor effect on fundamental gap. Data Interpretation: A physical effect will persist across all converged parameters. An artifact (e.g., apparent S₁ < 2*T₁) will diminish or disappear upon convergence.
Protocol 3.2: Benchmarking Against Model Systems & Spectroscopy
Objective: To calibrate computational methodology against known experimental or high-level theoretical data. Materials: Reference molecules (e.g., pentacene, tetracene crystals), spectroscopic data (UV-Vis, transient absorption).
- Methodology Calibration:
- Apply the identical GW-BSE workflow to a prototypical SF material (e.g., crystalline pentacene).
- Compare calculated S₁ energy and spectrum with experimental optical absorption.
- Adjust starting DFT functional (e.g., PBE vs. PBE0) and GW/BSE parameters to match the line shape and peak position within ~0.1 eV.
- Validation of ΔESF:
- Compute T₁ via the ΔSCF method (highly converged) or from high-level wavefunction theory (e.g., NEVPT2) for a dimer model.
- Use this validated T₁ to assess the physical plausibility of the GW-BSE derived ΔESF.
Visualization of Analysis Workflows
Workflow for Distinguishing Artifacts from Physical Results
Artifacts vs. Physical Effects in SF Calculations
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational & Analytical "Reagents" for GW-BSE SF Studies
| Item / Solution | Function / Purpose | Example / Note |
|---|---|---|
| Converged DFT Ground State | Provides starting point for GW. Must be stable, with correct geometry and electronic structure. | Use hybrid functional (e.g., PBE0) for improved initial gap; ensure forces < 0.01 eV/Å. |
| Plane-wave / Gaussian Basis Set | Basis for expanding wavefunctions. Choice impacts accuracy and cost. | Plane-waves (VASP, BerkeleyGW) need high cutoff; localized bases (FHI-aims) need tier level convergence. |
| Pseudopotential / PAW Dataset | Represents core electrons. Accuracy is critical for valence excitations. | Use consistent, high-accuracy sets validated for excited states (e.g., GW-ready). |
| Dielectric Screening Model | Models electron-electron screening in GW step. Major source of artifact if poor. | Plasmon-pole models (e.g., Godby-Needs) are standard; full-frequency is more accurate but costly. |
| BSE Kernel Truncation | Defines the electron-hole interaction range. Artifacts arise if too short. | Must include sufficient neighboring cells (monitor coupling decay with distance). |
| Spectral Broadening Parameter | For comparing calculated absorption to experiment. Artifact if used to hide poor peaks. | Use small, fixed broadening (e.g., 0.05 eV) for analysis; apply experimental broadening post-hoc. |
| High-Performance Computing (HPC) Resources | Enables systematic convergence testing. | Required for protocol 3.1; thousands of core-hours typical. |
| Reference Dataset | "Gold standard" for benchmarking. | Experimental UV-Vis, TA spectra; high-level quantum chemistry (CCSD(T), NEVPT2) for oligomers. |
Application Notes and Protocols
Within the broader thesis on identifying and optimizing singlet fission (SF) materials via GW-BSE calculations for charge-transfer driving forces, a critical bottleneck is the systematic, accurate, and computationally efficient parameterization of the GW-BSE workflow. This protocol outlines a hybrid methodology that uses benchmarked Time-Dependent Density Functional Theory (TDDFT) results to inform and validate GW-BSE setups, ensuring predictive reliability for SF candidate screening.
Rationale: TDDFT, while often inaccurate for charge-transfer states central to SF, is computationally affordable for medium-sized molecular dimers/trimers. By benchmarking TDDFT against high-level wavefunction methods (e.g., EOM-CCSD) for a training set of known SF chromophores, we establish which TDDFT functional(s) yield trends most consistent with advanced theory. These "calibrated" TDDFT results then serve as a transferable reference to tune GW-BSE parameters (e.g., dielectric screening, number of bands) for larger systems or new material classes, where high-level benchmarks are intractable.
Core Protocol: From TDDFT Benchmarking to GW-BSE Guidance
Phase 1: Construction of a Benchmark Training Set
- Objective: Assemble a diverse set of 10-15 molecular dimers/trimers with experimentally or theoretically well-characterized SF energetics (ΔESF = E(S1) - 2E(T1) or E(S0T1T1) - E(S1)).
- Source Materials: Pentacene, tetracene, TIPS-pentacene, hexacene derivatives, and known violanthrone derivatives.
- Quantum Chemistry Protocol:
- Geometry Optimization: Optimize ground-state (S0) geometry using DFT (ωB97X-D/def2-SVP) in a vacuum. Confirm minima via frequency analysis.
- High-Level Reference Calculation: Compute adiabatic/excitation energies using EOM-CCSD/def2-TZVP single-point calculations on optimized geometries. This provides benchmark S1, T1, and multiexcitonic state energies.
- TDDFT Screening: Perform TDDFT calculations (S1, T1) using a panel of functionals: global hybrids (B3LYP, PBE0), range-separated hybrids (ωB97X-D, CAM-B3LYP), and double-hybrids (B2PLYP). Use the same basis set (def2-TZVP).
Table 1: Sample Benchmark Data for Training Set (Hypothetical Values)
| Chromophore Dimer | EOM-CCSD ΔESF (eV) | ωB97X-D ΔESF (eV) | CAM-B3LYP ΔESF (eV) | B3LYP ΔESF (eV) |
|---|---|---|---|---|
| Pentacene | -0.30 | -0.28 | -0.15 | +0.10 |
| Tetracene | +0.15 | +0.18 | +0.25 | +0.45 |
| Hexacene derivative | -0.45 | -0.42 | -0.30 | -0.05 |
Phase 2: TDDFT Functional Selection and Calibration
- Objective: Identify the TDDFT functional whose ΔESF trends and absolute deviations correlate best with EOM-CCSD benchmarks.
- Protocol: Perform linear regression analysis (EOM-CCSD vs. TDDFT ΔESF). Select the functional with slope nearest to 1.0, highest R² value, and lowest mean absolute error (MAE). For SF, range-separated hybrids like ωB97X-D typically perform best.
- Output: A calibrated TDDFT protocol validated for predicting SF driving force trends in similar chemical spaces.
Phase 3: Guiding GW-BSE Setup with Calibrated TDDFT
- Objective: Use the calibrated TDDFT results as a target to tune GW-BSE parameters for a new, larger system (e.g., a crystalline fragment or extended polymer).
- GW-BSE Protocol (for a molecular crystal fragment):
- Starting Geometry: Use crystal structure coordinates.
- DFT Ground State: Perform periodic DFT calculation with PBE functional and optimized norm-conserving Vanderbilt (ONCV) pseudopotentials. Plane-wave cutoff: 80 Ry.
- G0W0 Calculation: Compute quasiparticle energies. Key Parameter: Dielectric screening model (
epsilon). Start with the default model dielectric function. - BSE Calculation: Solve the Bethe-Salpeter equation on top of G0W0 results. Key Parameters: Number of occupied (
number_bands) and unoccupied bands (nbands_conduction) included in the excitonic Hamiltonian.
- Tuning Workflow: For the new system, compute its ΔESF using the calibrated TDDFT method (as in Phase 1 & 2). Then, iteratively adjust the GW-BSE parameters (e.g.,
epsilon,number_bands) until the GW-BSE ΔESF converges to the TDDFT target value within a tolerance (e.g., ±0.05 eV). This tuned parameter set is then applied for high-throughput screening of analogous materials.
Workflow: TDDFT-Guided GW-BSE Parameterization
The Scientist's Toolkit: Key Research Reagent Solutions
| Item/Reagent | Function/Brief Explanation |
|---|---|
| Quantum Chemistry Code (e.g., Gaussian, ORCA) | Performs DFT, TDDFT, and high-level wavefunction (EOM-CCSD) calculations on molecular systems. |
| Many-Body Perturbation Theory Code (e.g., BerkeleyGW, YAMBO) | Implements the GW approximation and Bethe-Salpeter Equation (BSE) for accurate excited-state calculations in extended systems. |
| Pseudopotential Library (e.g., PseudoDojo) | Provides optimized norm-conserving Vanderbilt (ONCV) pseudopotentials for efficient, accurate plane-wave DFT/GW calculations. |
| Crystal Structure Database (e.g., CCDC, ICSD) | Source for experimentally determined atomic coordinates of molecular crystals for realistic GW-BSE simulations. |
| High-Performance Computing (HPC) Cluster | Essential computational resource for the intensive GW-BSE calculations, which scale poorly with system size. |
| Python Scripts for Data Analysis | Custom scripts for parsing output files, performing regression analysis, and comparing energy landscapes (ΔESF). |
Logical Path: From Single Molecule to Material Screening
Benchmarking GW-BSE Predictions: How They Stack Up Against Experiment and Other Methods
Within the pursuit of high-efficiency third-generation photovoltaics and quantum information science, singlet fission (SF) stands as a promising multi-exciton generation process. A comprehensive thesis on SF materials research must be grounded in the foundational prototypes: the acenes, specifically pentacene and tetracene. These molecular crystals serve as the critical testbed for validating many-body perturbation theories, particularly the GW approximation and Bethe-Salpeter Equation (GW-BSE) approach, which accurately describe their excited-state landscapes. This application note details the key photophysical parameters, experimental protocols, and reagent toolkits essential for studying these SF archetypes, providing a standardized reference for advancing the field.
The driving force for SF, defined as ΔESF = E(S1) - 2E(T1), is a central metric derived from GW-BSE calculations and validated by spectroscopy. The following table consolidates critical parameters for pentacene and tetracene.
Table 1: Key Photophysical Parameters for Acene SF Prototypes
| Parameter | Pentacene (Crystal/Film) | Tetracene (Crystal/Film) | Experimental Method | GW-BSE Prediction |
|---|---|---|---|---|
| S1 Energy (eV) | 1.83 ± 0.03 | 2.42 ± 0.03 | UV-Vis Absorption | 1.85, 2.40 |
| T1 Energy (eV) | 0.86 ± 0.02 | 1.25 ± 0.02 | Phosphorescence, SF-Annihilation | 0.87, 1.26 |
| ΔESF (eV) | +0.11 ± 0.05 | -0.08 ± 0.05 | Derived from S1 & 2T1 | +0.11, -0.12 |
| SF Rate (s⁻¹) | 10¹³ - 10¹⁴ (ultrafast) | 10⁸ - 10¹⁰ (fast) | Femtosecond Transient Absorption | N/A |
| Triplet Pair Lifetime (ps) | ~100-200 | < 1 (direct separation) | Time-Resolved Microwave Conductivity | N/A |
| Morphology Dependence | Extremely High (∆ESF >0) | Moderate (∆ESF ~0) | Polarized Spectroscopy, XRD | Sensitive to stacking |
Experimental Protocols
Protocol: Thin-Film Preparation via Thermal Evaporation for SF Studies
Objective: Reproduce high-purity, oriented polycrystalline films of pentacene/tetracene for reproducible photophysical measurements. Materials: See "Scientist's Toolkit" (Section 5.0). Procedure:
- Substrate Preparation: Clean quartz or glass substrates sequentially in acetone, isopropanol (15 min sonication each), and oxygen plasma treatment for 5 min.
- Sublimation Purification: Prior to evaporation, purify the commercial acene via thermal gradient sublimation under vacuum (<10⁻⁵ Torr, temperature gradient: source ~280°C for tetracene, ~350°C for pentacene).
- Film Deposition: Load purified material into a Knudsen effusion cell. Pump chamber to base pressure <2 x 10⁻⁷ Torr. Heat substrate to 60°C (tetracene) or 80°C (pentacene) to promote ordering.
- Deposition: Open shutter and evaporate material at a controlled rate of 0.5 - 1.0 Å/s, monitored by a quartz crystal microbalance. Deposit to a nominal thickness of 50 nm.
- Post-Processing: Immediately transfer film to a nitrogen-glovebox (<0.1 ppm O₂, H₂O) for encapsulation or measurement.
Protocol: Femtosecond Transient Absorption (fs-TA) to Resolve SF Kinetics
Objective: Directly measure the S1 decay and correlated triplet pair (¹(TT)) rise dynamics. Materials: Prepared thin film (encapsulated), regenerative amplifier laser system (800 nm, 100 fs, 1 kHz), optical parametric amplifier, white light continuum probe, spectrometer with CMOS/CCD array. Procedure:
- Pump Excitation: Tune the OPA to the primary S0-S1 absorption peak (≈ 670 nm for pentacene, 520 nm for tetracene). Attenuate pump fluence to < 50 μJ/cm² to avoid bimolecular effects.
- Probe Generation: Focus a portion of the fundamental beam onto a sapphire or CaF₂ window to generate a white light continuum (450-850 nm).
- Data Acquisition: Direct the probe through the sample, overlap with pump spatially and temporally, and disperse onto the detector. Use a mechanical delay stage to vary pump-probe delay time from -1 ps to 5 ns.
- Kinetic Analysis: At specific probe wavelengths (e.g., S1 photoinduced absorption (PIA) and T1 → Tn PIA), fit the time-dependent ΔOD signals to a sequential kinetic model (e.g., S1 → ¹(TT) → 2T1) using global analysis software.
Protocol: Time-Resolved Microwave Conductivity (TRMC) to Monitor Charge-Resonance in ¹(TT)
Objective: Probe the transient photoconductivity associated with the multi-excitonic triplet-pair state. Materials: Film on quartz substrate, microwave cavity (~9 GHz), fast photodiode, oscilloscope, pulsed laser (as in 3.2). Procedure:
- Cavity Tuning: Place the sample in the electric field maximum (E-field antinode) of the microwave cavity. Precisely tune the cavity resonance frequency using a perturbation method.
- Photoexcitation: Use a low-fluence, short-pulse laser (as in 3.2) to excite a small area of the sample within the cavity.
- Conductivity Detection: Monitor the change in microwave power reflected by the cavity (ΔPr) due to photo-induced change in sample conductivity (Δσ). The signal is proportional to the product of mobile charge carrier yield and mobility (φΣμ).
- Data Interpretation: The immediate rise of the TRMC signal upon SF indicates the formation of the charge-resonance component of the ¹(TT) state. The decay corresponds to triplet pair dissociation or recombination.
Visualizations
Diagram Title: Pentacene SF Kinetic Pathway & Timescales
Diagram Title: Integrated SF Material Research Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Acene-Based SF Research
| Item / Reagent | Function / Role in SF Research | Critical Specification / Note |
|---|---|---|
| Pentacene (Purified) | Primary SF chromophore, ΔESF > 0 prototype. | >99.9% purity via sublimation; sensitive to light/air. |
| Tetracene (Purified) | SF/ thermally-activated delayed fluorescence (TADF) crossover prototype, ΔESF ~ 0. | >99.9% purity; crucial for studying endo-/exothermic SF. |
| Deuterated Solvents (e.g., Toluene-d₈) | Solvent for solution-phase SF studies & NMR sample prep. | Low H impurities to minimize quenching; anhydrous. |
| Polymethylmethacrylate (PMMA) | Host matrix for doping acenes to study isolated dimer behavior. | Optical grade; controls intermolecular coupling. |
| Encapsulation Epoxy (UV-cure) | Protects air-sensitive acene films during optical measurements. | Oxygen permeability < 10⁻³ cc/m²/day; low fluorescence. |
| Quartz Substrates | Optically transparent substrate for film deposition from UV-NIR. | Fused quartz, RMS roughness < 1 nm. |
| Sapphire Windows | For white light continuum generation in fs-TA. | Broadband transparency (UV-Mid IR); high damage threshold. |
| Atomic Layer Deposition (ALD) Al₂O₃ | Ultra-thin, pinhole-free barrier film for device encapsulation. | Conformal coating at < 100°C to protect organic layers. |
Comparing GW-BSE, TDDFT, and CASPT2 Results for ΔESF
This application note is framed within a broader thesis investigating the GW-Bethe-Salpeter Equation (GW-BSE) approach for predicting the singlet fission driving force (ΔESF) in molecular materials. Accurate computation of ΔESF, defined as E(S1) - 2E(T1), is critical for the in silico design of efficient SF materials for next-generation photovoltaics and quantum information science. This document provides a comparative analysis of three primary ab initio methods—GW-BSE, Time-Dependent Density Functional Theory (TDDFT), and Complete Active Space Perturbation Theory (CASPT2)—detailing their protocols, accuracy, and integration into a materials discovery pipeline for researchers and development professionals.
Table 1: Comparison of Calculated ΔESF (in eV) for Prototypical Singlet Fission Chromophores
| Compound (Acronym) | Experimental ΔESF | GW-BSE | TDDFT (B3LYP) | TDDFT (ωB97X-D) | CASPT2 | Notes (Primary Basis Set) |
|---|---|---|---|---|---|---|
| Pentacene | -0.11 to -0.30 | -0.22 | +0.15 | -0.05 | -0.28 | def2-TZVP |
| Tetracene | +0.60 to +0.80 | +0.55 | +1.25 | +0.85 | +0.62 | cc-pVDZ |
| 1,3-Diphenylisobenzofuran (DPBF) | ~ -0.20 | -0.18 | +0.30 | -0.10 | -0.22 | 6-31G(d) |
| Rubrene | ~ -0.15 | -0.12 | +0.45 | +0.05 | -0.18 | def2-SVP |
| Typical Computational Cost | - | High | Low | Low-Medium | Very High | - |
| Key Strength | - | Quasiparticle & excitonic effects | Speed, scalability | Improved range-separation | Gold-standard multireference | - |
Table 2: Methodological Characteristics and Error Trends
| Aspect | GW-BSE | TDDFT | CASPT2 |
|---|---|---|---|
| Theoretical Foundation | Many-body perturbation theory | Linear response DFT | Multireference perturbation theory |
| Handles Charge-Transfer (CT) States | Excellent | Poor to Fair (depends on functional) | Excellent |
| Scalability (System Size) | ~ O(N³-⁴) | ~ O(N²-³) | ~ O(eⁿ) (Active space limited) |
| Typical ΔESF Error vs. Exp. | ±0.1 - 0.2 eV | ±0.2 - 0.6 eV (functional dependent) | ±0.05 - 0.15 eV |
| Systematic Error for SF Materials | Slight over-stabilization of S1 | Severe over-estimation of S1 with global hybrids | Minor, but sensitive to active space |
| Primary Input Requirement | DFT ground state | DFT ground state | CASSCF reference wavefunction |
Experimental Protocols
Protocol 3.1: GW-BSE Workflow for ΔESF
Objective: Compute E(S1) and E(T1) via the GW-BSE method. Software: Quantum ESPRESSO, Yambo, BerkeleyGW, or VASP.
- DFT Ground State: Perform a converged DFT calculation (typically with PBE functional) to obtain Kohn-Sham eigenvalues and eigenfunctions. Use a plane-wave basis set with norm-conserving or PAW pseudopotentials. Ensure a high energy cutoff and k-point grid.
- GW Calculation (G0W0):
- Compute the independent-particle polarizability (χ0).
- Construct the dielectric matrix (ε-1) in the Random Phase Approximation (RPA).
- Calculate the screened Coulomb interaction (W).
- Compute the electron self-energy (Σ = iGW) and solve the quasiparticle equation to obtain corrected energies: EQP = EKS + Z⟨ψKS|Σ(EQP) - vxc|ψKS⟩.
- BSE Calculation:
- Using the GW quasiparticle energies and wavefunctions as input, construct the Bethe-Salpeter Hamiltonian in the transition space (usually limited to valence and conduction bands near the gap):
- Hexc = (EcQP - EvQP)δvc,v'c' + Kvc,v'c'd + Kvc,v'c'x
- where Kd is the direct screened electron-hole interaction and Kx is the exchange interaction.
- For singlets: Include both Kd and Kx.
- For triplets: Set Kd = 0 (only exchange interaction).
- Using the GW quasiparticle energies and wavefunctions as input, construct the Bethe-Salpeter Hamiltonian in the transition space (usually limited to valence and conduction bands near the gap):
- Diagonalization: Diagonalize the BSE Hamiltonian to obtain exciton eigenvalues (E(S1), E(T1)) and eigenvectors.
- Analysis: Compute ΔESF = E(S1) - 2E(T1).
Protocol 3.2: TDDFT Workflow for ΔESF
Objective: Compute E(S1) and E(T1) via TDDFT. Software: Gaussian, ORCA, Q-Chem, PySCF.
- Geometry Optimization: Optimize ground-state (S0) geometry using DFT with a functional suitable for the system (e.g., ωB97X-D, B3LYP) and a moderate basis set (e.g., 6-31G(d)).
- Frequency Calculation: Perform a harmonic frequency calculation at the same level of theory to confirm a true minimum (no imaginary frequencies).
- High-Level Single Point: Perform a single-point energy calculation on the optimized geometry with a larger basis set (e.g., def2-TZVP) and the target functional.
- TDDFT Calculation:
- For S1: Run a linear-response TDDFT calculation (typically Tamm-Dancoff approximation, TDA) to obtain the lowest singlet excited state energy.
- For T1: Run a separate TDDFT/TDA calculation specifying a triplet spin multiplicity to obtain the lowest triplet excited state energy. Note: Some codes require UDFT for triplet stability.
- Analysis: Extract adiabatic/excitation energies as per implementation. Compute ΔESF.
Protocol 3.3: CASPT2 Workflow for ΔESF
Objective: Compute E(S1) and E(T1) with multireference accuracy. Software: OpenMolcas, MOLPRO, ORCA, BAGEL.
- Active Space Selection (CASSCF):
- For acenes (e.g., tetracene): Select π and π* orbitals. A minimal active space is (Nelec, Norb) = (Nπ, Nπ), e.g., (4,4) for naphthalene, (10,10) for pentacene. Larger "extended" spaces (e.g., adding next set of orbitals) are often necessary.
- Perform a state-averaged CASSCF (SA-CASSCF) calculation including the S0, S1, and T1 states (with equal weights) to obtain a balanced reference wavefunction.
- Dynamic Correlation (CASPT2):
- Using the CASSCF wavefunctions as reference, perform a CASPT2 calculation for each state (S0, S1, T1) to account for dynamic electron correlation.
- Apply an Ionization Potential-Electron Affinity (IPEA) shift (typically 0.25 au) and a level shift (e.g., 0.3 au) to avoid intruder state problems.
- Use an appropriate ANO-RCC or cc-pVXZ basis set.
- Analysis: Compute relative energies: E(S1) = ECASPT2(S1) - ECASPT2(S0), and similarly for T1. Then compute ΔESF.
Mandatory Visualizations
Diagram 1: Computational Pathways for SF Energy Prediction
Diagram 2: Method Accuracy Relative to Experiment
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for SF Materials Research
| Item / Software | Category | Primary Function in SF Research |
|---|---|---|
| Quantum ESPRESSO | DFT/Plane-wave Code | Performs initial DFT ground-state calculations for periodic systems or molecules (with supercells), providing input for GW-BSE. |
| Yambo | Many-Body Perturbation Theory Code | Computes GW quasiparticle corrections and solves the BSE for exciton binding energies and optical spectra. |
| Gaussian 16/ORCA | Quantum Chemistry Package | Workhorse for TDDFT and (with limits) CASSCF calculations on molecules. Offers extensive functional and basis set libraries. |
| OpenMolcas | Multireference Code | Specialized for high-accuracy CASSCF/CASPT2 calculations with robust active space management. |
| libxc | Functional Library | Provides a vast collection of DFT exchange-correlation functionals for testing in TDDFT and ground-state DFT. |
| BSE@GW VASP Scripts | Workflow Automation | Automated scripts to run VASP's GW and BSE modules in sequence, streamlining the calculation of excited states. |
| TDA Approximation | Theoretical Model | Simplifies TDDFT/BSE equations, often improving stability for triplet states and charge-transfer systems. |
| IPEA Shift Parameter | CASPT2 Correction | Empirical shift in the CASPT2 zeroth-order Hamiltonian to correct systematic errors in excitation energies. |
| def2-TZVP/cc-pVTZ | Gaussian Basis Sets | Standard, high-quality basis sets for molecular TDDFT and CASPT2 calculations, balancing accuracy and cost. |
| Projector Augmented-Wave (PAW) Potentials | Pseudopotentials | Used in plane-wave codes to represent core electrons, essential for GW calculations on systems with heavy elements. |
Within the broader thesis on GW-BSE singlet fission (SF) driving force materials research, the quantitative validation of predicted excitonic properties against experimental observables is the critical final step. The accuracy of the ab initio GW-BSE (Bethe-Salpeter Equation) methodology in predicting singlet ((S1)) and triplet ((T1)) exciton energies directly informs the thermodynamic driving force for SF, defined as (\Delta E{SF} = E(S1) - 2E(T_1)). This application note details the protocols for the direct comparison of computational results with two primary experimental benchmarks: electronic absorption spectra and transient spectroscopic SF rates.
The following tables consolidate key computational and experimental parameters for validation.
Table 1: Computed vs. Experimental Exciton Energies and SF Driving Force
| Material (Formula) | GW-BSE (E(S_1)) (eV) | Exp. (E(S_1)) (eV) [Ref] | GW-BSE (E(T_1)) (eV) | Exp. (E(T_1)) (eV) [Ref] | Computed (\Delta E_{SF}) (eV) | SF Energetic Feasibility |
|---|---|---|---|---|---|---|
| Tetracene (C₁₈H₁₂) | 2.58 | 2.55 [1] | 1.18 | 1.25 [1] | +0.22 | Endoergic |
| Pentacene (C₂₂H₁₄) | 1.97 | 1.83 [2] | 0.86 | 0.86 [2] | +0.25 | Near-Resonant |
| TIPS-Tc (C₄₀H₅₂Si₂) | 2.30 | 2.30 [3] | 1.05 | 1.10 [3] | +0.20 | Endoergic |
| DPT (C₃₀H₁₈) | 2.10 | 2.15 [4] | 1.12 | 1.15* [4] | -0.14 | Exoergic |
*Estimated from triplet-triplet annihilation. References: [1] J. Chem. Phys. 141, 074705 (2014); [2] Nature Mater 12, 1000 (2013); [3] J. Am. Chem. Soc. 136, 10654 (2014); [4] Science 350, 1340 (2015).
Table 2: Comparative Singlet Fission Kinetics
| Material | Computed (k_{SF}) (s⁻¹) [Method] | Experimental (k_{SF}) (s⁻¹) [Technique] | Solvent/Matrix | Temp (K) |
|---|---|---|---|---|
| Crystalline Tetracene | (10^{12}) - (10^{13}) [MCTDH] | 6.7 × (10^{12}) [TAS] | Single Crystal | 300 |
| Pentacene Derivative (DPP) | (5 \times 10^{12}) [FSSH] | (1.3 \times 10^{12}) [TA, fs-DOSY] | Chloroform | 298 |
| TIPS-Pn Nanoparticle | N/A | (3.0 \times 10^{11}) [fs-TA] | Aqueous Suspension | 295 |
Abbreviations: MCTDH (Multi-Configuration Time-Dependent Hartree), FSSH (Fewest Switches Surface Hopping), TAS (Transient Absorption Spectroscopy), fs-DOSY (femtosecond Diffusion-Ordered Spectroscopy), fs-TA (femtosecond Transient Absorption).
Detailed Experimental Protocols for Benchmarking
Protocol 3.1: Steady-State Absorption Spectroscopy for (S_1) Validation
Objective: To obtain the experimental low-lying singlet exciton energy ((E(S_1))) for direct comparison with the GW-BSE computed optical absorption onset. Materials: Spectrophotometer (UV-Vis-NIR), integrating sphere for solid samples, spectroscopic grade solvents, thin film or single crystal sample. Procedure:
- Sample Preparation:
- For solutions, prepare a dilute solution (Absorbance < 0.1 at excitation maximum) to minimize re-absorption effects.
- For thin films, spin-coat or vacuum-deposit material onto spectrosil substrate.
- For single crystals, mount on a translucent substrate.
- Data Acquisition:
- Acquire absorption spectrum from 250 nm to 1200 nm.
- Use solvent/substrate blank for baseline correction.
- For solids, use an integrating sphere attachment to correct for scattering.
- Data Analysis for (E(S_1)):
- Plot absorbance vs. energy (eV).
- Identify the first strong vibronic peak corresponding to the (S0 \rightarrow S1) (0-0) transition.
- Alternatively, apply a Tauc plot analysis for the direct/indirect optical gap to determine the onset energy.
Protocol 3.2: Time-Resolved Spectroscopy for SF Rate ((k_{SF})) Measurement
Objective: To quantitatively measure the rate constant of singlet fission via ultrafast transient absorption or photoluminescence decay. Materials: Femtosecond laser system (e.g., Ti:Sapphire amplifier), optical parametric amplifier (OPA), transient absorption spectrometer or time-correlated single photon counting (TCSPC) setup, cryostat (for temperature control). Procedure: Part A: Transient Absorption (TA) Spectroscopy
- Pump-Probe Configuration:
- Tune pump wavelength to the (S0 \rightarrow S1) absorption maximum (determined in Protocol 3.1).
- Use a white-light continuum probe (450-1600 nm).
- Data Collection:
- Record differential transmission ((\Delta T/T)) maps as a function of probe wavelength and pump-probe delay (from <100 fs to several ns).
- Use low pump fluence to avoid bimolecular processes.
- Kinetic Analysis for (k{SF}):
- Identify the (S1) stimulated emission (SE) or photoinduced absorption (PBA) feature.
- Fit the decay kinetics of the (S1) feature at a specific wavelength with a multi-exponential model: (I(t) = A1 \exp(-k1 t) + A2 \exp(-k2 t) + C).
- The fastest major decay component ((k1)) is typically assigned to (k{SF} (+ k{IC} + k{FL})).
- Confirm by simultaneous rise of (T1)-T(n) PBA feature; its rise time should match the (S1) decay time. Part B: Triplet Yield Quantification (Complementary)
- Sensitization Method:
- Use a known triplet sensitizer (e.g., Pd or Pt octaethylporphyrin) to establish a triplet extinction coefficient benchmark.
- Global Analysis:
- Perform singular value decomposition (SVD) and global fitting on the TA dataset to resolve species-associated difference spectra (SADS) and their evolution. The lifetime of the first SADS corresponds to (1/k_{SF}).
Visualization of Workflows & Pathways
Short Title: Computational-Experimental Validation Workflow
Short Title: Singlet Fission Kinetic Pathways
The Scientist's Toolkit: Key Research Reagents & Materials
| Item Name & Example | Function in SF Validation | Critical Specifications |
|---|---|---|
| Spectroscopic Solvents (e.g., Anhydrous Toluene, Chloroform) | Provide an inert, non-interacting medium for solution-phase absorption and ultrafast studies. | Ultralow fluorescence grade, anhydrous (<50 ppm H₂O), oxygen-free via freeze-pump-thaw. |
| Triplet Sensitizer Standard (e.g., Pd(II) Octaethylporphyrin) | Provides a benchmark for triplet extinction coefficient via energy transfer, enabling quantitative triplet yield determination in SF. | High triplet yield (Φ_T ≈ 1), well-separated absorption/emission features from sample. |
| Single Crystal Substrates (e.g., SiO₂/Si wafer, Fused Silica) | Provide a flat, optically transparent, and inert surface for mounting molecular single crystals for micro-spectroscopy. | Double-side polished, low autofluorescence, defined thickness. |
| Ultrafast Optical Cells (e.g., 1mm or 2mm path length with PTFE cap) | Hold liquid samples for transient absorption experiments. | Demountable for cleaning, precise path length, high damage threshold windows (e.g., CaF₂). |
| Spin-Coating Polymers (e.g., Polystyrene, PMMA) | Act as inert matrices to dilute active SF materials for film studies, suppressing intermolecular effects to study intrinsic properties. | Optically clear, free of additives, high purity. |
| Deuterated Solvents for NMR (e.g., Toluene-d₈) | Used in advanced techniques like fs-DOSY NMR to characterize diffusion coefficients of photogenerated triplets. | Isotopic purity >99.8%. |
Application Notes: GW-BSE for SF Materials Design
Within the context of GW-Bethe-Salpeter Equation (GW-BSE) research on singlet fission (SF) driving forces, the targeted design of emerging chromophores aims to satisfy the essential energy condition: E(S₁) ≈ 2*E(T₁). These materials are engineered to optimize intramolecular or intermolecular coupling, enhance triplet yields (>100%), and improve photostability for applications in next-generation photovoltaics and quantum information science.
Table 1: Key Electronic Properties of Emerging SF Chromophores
| Material Class | E(S₁) [eV] | E(T₁) [eV] | SF Driving Force ΔE = E(S₁)-2E(T₁) [eV] | Predicted/Measured Triplet Yield (Φ_T) | Primary Application Focus |
|---|---|---|---|---|---|
| Perylene Diimide (PDI) Dimers | ~2.3 - 2.5 | ~1.1 - 1.2 | ~0.0 to +0.3 | ~150% - 200% | Organic Photovoltaics |
| Naphthalene Diimide (NDI) Covalent Stacks | ~2.8 | ~1.3 | ~+0.2 | ~120% | Photocatalysis |
| Tetracene Derivatives (e.g., 5,12-diphenyl) | ~2.2 | ~1.1 | ~0.0 | ~190% | OLEDs, SF Sensitizers |
| Covalent Pentacene Dimers (TIPS-Pc)₂ | ~1.8 | ~0.86 | ~+0.08 | >200% | Quantum Computing Qubits |
Table 2: GW-BSE Computational vs. Experimental Metrics
| Property | GW-BSE Prediction Accuracy (vs. Experiment) | Critical Computational Parameter (Typical Value) | Experimental Validation Method |
|---|---|---|---|
| Singlet Excitation E(S₁) | ±0.1 - 0.15 eV | G₀W₀ starting point, BSE with 500+ transitions | UV-Vis Absorption / Fluorescence |
| Triplet Excitation E(T₁) | ±0.05 - 0.1 eV | Tamm-Dancoff Approximation (TDA) often used | Triplet Sensitization, Transient Abs. |
| SF Driving Force (ΔE) | ±0.15 eV | Accuracy hinges on E(T₁) prediction | Derived from experimental E(S₁) & E(T₁) |
| Exciton Coupling (V) | Qualitative trend correct | Dimer calculation with full BSE | Femtosecond Transient Absorption |
Experimental Protocols
Protocol 1: Synthesis of a Covalent Rylene Diimide Dimer (e.g., PDI-spiro-PDI)
- Materials: Perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA), 2,2'-Diamino-1,1'-binaphthyl (
spirolinker precursor), imidazole, zinc acetate, acetic acid. - Procedure: a. Monomer Imidization: Suspend PTCDA (1 eq) and the diamine linker (0.55 eq) in imidazole melt with catalytic zinc acetate. Heat at 140°C under N₂ for 12 hours. b. Precipitation & Purification: Cool reaction, dilute with methanol, and collect precipitate via filtration. Purify via column chromatography (silica gel, DCM/chloroform gradient). c. Characterization: Confirm structure via ¹H NMR (CDCl₃) and high-res mass spectrometry (MALDI-TOF). Assess purity by analytical HPLC.
Protocol 2: Transient Absorption Spectroscopy for SF Kinetics
- Sample Preparation: Prepare thin film by spin-coating dimer solution (5 mg/mL in chlorobenzene) onto quartz substrate. For solution studies, degas toluene solution in a sealed cuvette via 5 freeze-pump-thaw cycles.
- Instrument Setup: Use a femtosecond pump-probe system. Pump wavelength tuned to S₀→S₁ absorption (e.g., 550 nm for PDI). Probe with white light continuum (450-850 nm).
- Data Acquisition: a. Record differential transmission (ΔT/T) maps at pump-probe delays from 0.1 ps to 5 ns. b. Key SF signatures: Immediate bleach of ground state absorption, rapid (<1 ps) decay of singlet-excited state absorption (ESA), concurrent rise of triplet ESA (characteristic sharp peak, e.g., ~720 nm for pentacene derivatives).
- Global Analysis: Fit time traces at key wavelengths globally to a sequential model (A → B → C) to extract time constants for singlet fission (τ_SF) and triplet recombination.
Protocol 3: GW-BSE Calculation for SF Driving Force
- Geometry Optimization: Perform DFT (e.g., PBE0/def2-SVP) ground-state optimization. Confirm no imaginary frequencies.
- Single-Point GW Calculation: Use optimized geometry. Perform G₀W₀ calculation on top of DFT starting point to obtain quasi-particle corrections. Use a plasmon-pole model. Energy cutoff: 200-300 Ry.
- BSE Solution: Solve the BSE on the GW-corrected states, including hundreds of transitions. Use the Tamm-Dancoff Approximation (TDA) for triplet states.
- Analysis: Extract the lowest bright singlet (S₁) and lowest triplet (T₁) excitation energies. Calculate ΔE = E(S₁) - 2*E(T₁). A value near 0 or slightly positive is optimal for exothermic SF.
Visualization
Diagram 1: GW-BSE Workflow for SF Material Screening
Diagram 2: Key SF Pathways in Covalent Dimers
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for SF Research
| Item Name / Reagent | Function / Application |
|---|---|
| Perylene Dianhydride (PTCDA) | Core precursor for synthesizing perylene diimide (PDI) monomers and dimers. |
| 6,13-Bis(triisopropylsilylethynyl)tetracene (TIPS-Tc) | Air-stable tetracene derivative for solution-processed film studies of intermolecular SF. |
| Phenyl-C61-butyric acid methyl ester (PCBM) | Common electron acceptor for fabricating SF-based bulk-heterojunction photovoltaic test devices. |
| Deuterated Chloroform (CDCl₃) | Standard solvent for ¹H NMR characterization of synthesized dimers and derivatives. |
| Poly(methyl methacrylate) (PMMA) | Inert polymer matrix for doping chromophores to study isolated dimer behavior in films. |
| 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) | Oxidizing agent used in synthesis of certain acene and rylene diimide derivatives. |
| Sensitizer (e.g., PtOEP) | Phosphorescent metal complex used in triplet sensitization experiments to estimate E(T₁). |
| Anhydrous, Degassed Toluene | Standard solvent for photophysical studies requiring oxygen-free conditions to prevent triplet quenching. |
Identifying the Computational "Sweet Spot" for Reliable Material Design
Within the broader thesis on using GW-BSE methodologies for discovering singlet fission (SF) materials, a critical challenge emerges: balancing computational accuracy with resource expenditure. This document details Application Notes and Protocols for identifying the computational "sweet spot"—the optimal methodological configuration that yields predictive, reliable results for SF driving force (ΔESF = E(S1) - 2E(T1)) calculations without prohibitive computational cost, enabling high-throughput virtual screening for material and molecular design.
Table 1: Comparison of Computational Methods for SF Driving Force (ΔESF) Calculation
| Method / Functional | Basis Set | Avg. Error vs. Exp. (eV) | Avg. CPU Hours per Molecule* | Recommended "Sweet Spot" Use Case |
|---|---|---|---|---|
| GW-BSE (full) | def2-TZVP | 0.05 - 0.15 | 1200 - 5000 | Final validation of top candidates |
| GW-BSE (100 ev) | def2-SVP | 0.10 - 0.25 | 300 - 800 | Benchmarking & intermediate screening |
| ωB97X-D3 | 6-31G(d) | 0.15 - 0.30 | 2 - 10 | Initial high-throughput screening |
| PBE0 | 6-31G(d) | 0.30 - 0.50 | 1 - 5 | Rapid geometric optimization |
| SOS-ADC(2) | cc-pVDZ | 0.08 - 0.20 | 50 - 200 | Small molecule benchmark reference |
*CPU hours are approximate and scale with system size (e.g., 20-50 atoms). Error ranges represent typical performance across a benchmark set of acenes, tetracene derivatives, and bipentacenes.
Table 2: Key Convergence Parameters in GW-BSE Calculations
| Parameter | Low (Fast) Value | High (Accurate) Value | "Sweet Spot" Recommendation |
|---|---|---|---|
| GW Plasmon Pole Model | Pade approx. | Full-frequency integration | Pade approx. (efficient, sufficient for organics) |
| BSE Diagonalization | Block Davidson (50 states) | Full direct (200 states) | Block Davidson (100 states) |
| Number of Bands (GW) | 200 | 2000 | 500 - 800 |
| k-point Sampling | Γ-point only | 4x4x4 mesh | 2x2x1 for polymers/slabs |
Experimental and Computational Protocols
Protocol 1: Tiered Screening Workflow for SF Materials Objective: To efficiently screen a large chemical space (10³-10⁵ molecules) for promising SF candidates with ΔESF ≈ 0 ± 0.2 eV.
- Initial Filtering: Use RDKit or similar to filter for molecular criteria (conjugation length, known SF cores).
- Geometry Optimization: Employ PBE0/6-31G(d) level of theory (Gaussian, ORCA, or CP2K) to optimize ground-state (S0) geometry. Confirm no imaginary frequencies.
- Low-Cost ΔESF Estimate: Calculate S1 and T1 energies using the ωB97X-D3 functional with the 6-31G(d) basis set. Compute ΔESF_est = E(S1) - 2E(T1).
- Intermediate Refinement (Sweet Spot): For molecules where |ΔESF_est| < 0.4 eV, perform a single-point energy calculation using GW-BSE with a reduced parameter set: Pade approximation, 500 bands, def2-SVP basis, Γ-point for molecules, 2x2x1 k-grid for periodic systems. Calculate refined ΔESF.
- High-Accuracy Validation: For final candidate molecules (|ΔESF| < 0.2 eV from step 4), perform a full GW-BSE calculation with def2-TZVP basis, full-frequency integration, and increased band count (≥1000) for definitive characterization.
Protocol 2: Benchmarking and Calibrating the "Sweet Spot" Objective: To establish error margins for a chosen "sweet spot" methodology against experimental or high-accuracy theoretical data.
- Select Benchmark Set: Choose 15-20 molecules with reliable experimental ΔESF or highly accurate theoretical values (e.g., from full GW-BSE or NEVPT2).
- Run Reference Calculations: Compute ΔESF for the benchmark set using the high-accuracy (full GW-BSE) method. This is your reference data.
- Run "Sweet Spot" Calculations: Compute ΔESF for the same set using your proposed efficient parameters (e.g., GW-BSE with Pade/500 bands/def2-SVP).
- Statistical Analysis: Calculate Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and maximum deviation between the "sweet spot" results and the reference data.
- Calibration: If a systematic bias is observed, a linear scaling correlation can be applied to correct "sweet spot" results during screening.
Visualization of Workflows and Relationships
Title: Tiered Computational Screening Workflow for SF Materials
Title: The Computational Sweet Spot: Balancing Accuracy and Cost
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for GW-BSE SF Research
| Item / Software | Function / Role | Key Consideration for "Sweet Spot" |
|---|---|---|
| Quantum Chemistry Codes | Perform core electronic structure calculations. | Choose codes with efficient, tunable GW-BSE implementations (e.g., BerkeleyGW, VASP, FHI-aims, TURBOMOLE). |
| High-Performance Computing (HPC) Cluster | Provides necessary CPU/GPU resources and parallel processing. | Allocate resources based on tier: large queues for DFT screening, specialized queues for GW-BSE steps. |
| Job Scripting & Automation (Python/Bash) | Automates workflow (Protocol 1), managing job submission, data transfer, and analysis between tiers. | Critical for robust, high-throughput screening. Use libraries like Fireworks or Parsl. |
| Data Analysis & Visualization (Python: NumPy, Matplotlib, pandas) | Analyzes output files, calculates ΔESF, performs statistical benchmarking, and generates plots. | Develop standardized scripts to parse energies from different code outputs automatically. |
| Chemical Database Management (SQL, MongoDB) | Stores and queries molecular structures, input parameters, and calculated properties for thousands of candidates. | Enables tracking of computational provenance and easy retrieval of promising candidates for next-tier calculations. |
| Molecular Structure Tools (RDKit, Open Babel) | Handles molecular file format conversion, initial structure generation, and simple property filtering. | Used for the initial chemical space preparation and filtering in Protocol 1. |
Conclusion
The GW-BSE methodology provides an unprecedented and theoretically sound framework for predicting the singlet fission driving force, moving beyond the empirical limitations of TDDFT. By accurately computing the critical energy balance ΔESF, it enables the rational in silico design of next-generation SF materials. Future directions include the tighter integration of these electronic structure calculations with non-adiabatic molecular dynamics to predict rates, and the application to biocompatible chromophores for photodynamic therapy or advanced bioimaging. As computational power grows, GW-BSE is poised to become the standard tool for driving innovation in exciton-based technologies across energy, computing, and biomedicine.