The Hohenberg-Kohn Theorems: A Complete Guide for Materials & Drug Discovery Researchers
This comprehensive guide demystifies the foundational Hohenberg-Kohn theorems of Density Functional Theory (DFT) for researchers and drug development professionals.
The Hohenberg-Kohn Theorems: A Complete Guide for Materials & Drug Discovery Researchers
Abstract
This comprehensive guide demystifies the foundational Hohenberg-Kohn theorems of Density Functional Theory (DFT) for researchers and drug development professionals. We start by exploring the core principles—why electron density is the key variable and its profound implications for simplifying quantum mechanics. We then detail the methodological bridge from theory to practical application, including the Kohn-Sham equations and modern exchange-correlation functionals. The guide tackles common computational challenges, accuracy trade-offs, and optimization strategies crucial for realistic simulations of molecules and materials. Finally, we validate DFT's power by comparing it to other quantum chemical methods, showcasing its unmatched scalability and utility in predicting molecular properties, protein-ligand interactions, and solid-state phenomena for biomedical and materials science.
Beyond the Wavefunction: How Hohenberg-Kohn Theorems Revolutionized Quantum Mechanics
The Hohenberg-Kohn theorems establish the foundation of Density Functional Theory (DFT), proving that the ground-state electron density uniquely determines all properties of a many-electron system. This revolution shifted focus from the intractable many-body wavefunction, (\Psi(\mathbf{r}1, \mathbf{r}2, ..., \mathbf{r}_N)), to the manageable three-dimensional density, (n(\mathbf{r})). However, the core quantum many-body problem—the exponential scaling of the wavefunction's complexity with particle number—remains the fundamental barrier that DFT and related methods seek to circumvent. This whitepaper details the scale of this intractability, modern methodological approaches, and the implications for fields like ab initio drug discovery.
The Exponential Scaling of Wavefunction Complexity
The many-body wavefunction for an N-electron system depends on 3N spatial coordinates (and N spin coordinates). Discretizing each spatial dimension into just m points leads to a computational mesh of size (m^{3N}).
Table 1: Scaling of Wavefunction Representation
| System (N electrons) | Spatial Coordinates | Approximate Degrees of Freedom (m=10) | Memory for Full Wavefunction (Double Precision) |
|---|---|---|---|
| H₂ (N=2) | 6 | (10^6) | ~8 MB |
| H₂O (N=10) | 30 | (10^{30}) | ~(10^{17}) YB (Physically Impossible) |
| Small Protein (N~500) | 1500 | (10^{1500}) | Beyond any conceivable storage |
This exponential scaling makes the explicit storage or direct manipulation of (\Psi) impossible for all but the smallest systems, a manifestation of the "curse of dimensionality."
Methodological Approaches to Bypass Intractability
Density Functional Theory (DFT)
DFT, grounded by Hohenberg-Kohn, uses the electron density (n(\mathbf{r})) (3D) as the fundamental variable. The Kohn-Sham equations introduce a fictitious system of non-interacting electrons that yield the same density, reducing the problem to solving N one-electron equations.
Experimental/Computational Protocol: Kohn-Sham DFT Calculation
- Input: Atomic species and positions.
- Initial Guess: Generate trial electron density (n_{initial}(\mathbf{r})).
- Construct Kohn-Sham Potential:
- Solve Kohn-Sham Equations:
- (\left[ -\frac{1}{2} \nabla^2 + v{KS}(\mathbf{r}) \right] \phii(\mathbf{r}) = \epsiloni \phii(\mathbf{r}))
- Obtain N lowest eigenstates (\phi_i).
- Compute New Density: (n{new}(\mathbf{r}) = \sum{i=1}^N |\phi_i(\mathbf{r})|^2).
- Self-Consistent Field (SCF) Cycle: Iterate steps 3-5 until (n{new}) and (n{old}) converge (e.g., energy change < (10^{-6}) Ha).
- Output: Total energy, forces, electronic eigenvalues, density.
Wavefunction-Based Post-Hartree-Fock Methods
For higher accuracy, methods build upon a Hartree-Fock reference wavefunction but scale severely.
Table 2: Comparative Analysis of Many-Body Methods
| Method | Fundamental Variable | Key Approximation | Computational Scaling | Typical Application Scope | ||
|---|---|---|---|---|---|---|
| Full CI | Many-body (\Psi) | None (Exact within basis) | Factorial ~N! | Tiny molecules (<10 e⁻) | ||
| Coupled Cluster (CCSD) | Correlated (\Psi) | Truncation at double excitations | (O(N^6)) | Medium molecules (<50 e⁻) | ||
| Quantum Monte Carlo | Sampled ( | \Psi | ^2) | Fixed-node constraint | (O(N^3)) - (O(N^4)) | Small clusters, solids |
| Kohn-Sham DFT (LDA) | Electron density (n(\mathbf{r})) | Local density approximation (LDA) | (O(N^3)) | Bulk materials, large molecules | ||
| Hybrid DFT (B3LYP) | Electron density (n(\mathbf{r})) | Empirical hybrid functional | (O(N^3)) - (O(N^4)) | Molecules, reaction barriers |
The Scientist's Toolkit: Research Reagent Solutions for Electronic Structure
Table 3: Essential Computational "Reagents" in Many-Body Simulations
| Item (Software/Code) | Function/Purpose | Example |
|---|---|---|
| Basis Set | Set of one-electron functions used to expand orbitals/density. Determines accuracy and cost. | Gaussian-type orbitals (6-31G, cc-pVDZ), Plane waves (cutoff energy). |
| Pseudopotential | Replaces core electrons with an effective potential, reducing required basis size. | Norm-conserving (ONCV), Ultrasoft, Projector-augmented wave (PAW). |
| Exchange-Correlation (XC) Functional | Approximates quantum mechanical exchange and correlation effects in DFT. | LDA, GGA (PBE), Meta-GGA (SCAN), Hybrid (B3LYP, HSE06). |
| SCF Solver | Algorithm to achieve self-consistency in DFT/HF calculations. | Direct minimization, Davidson diagonalization, Pulay mixing (DIIS). |
| Quantum Chemistry Package | Integrated software suite implementing the above methods. | VASP, Quantum ESPRESSO (Plane-wave DFT); Gaussian, PySCF (Molecular). |
Visualization of Conceptual and Workflow Relationships
Diagram Title: Many-Body Solution Pathways from H-K Theorems
Diagram Title: Kohn-Sham DFT Self-Consistent Cycle
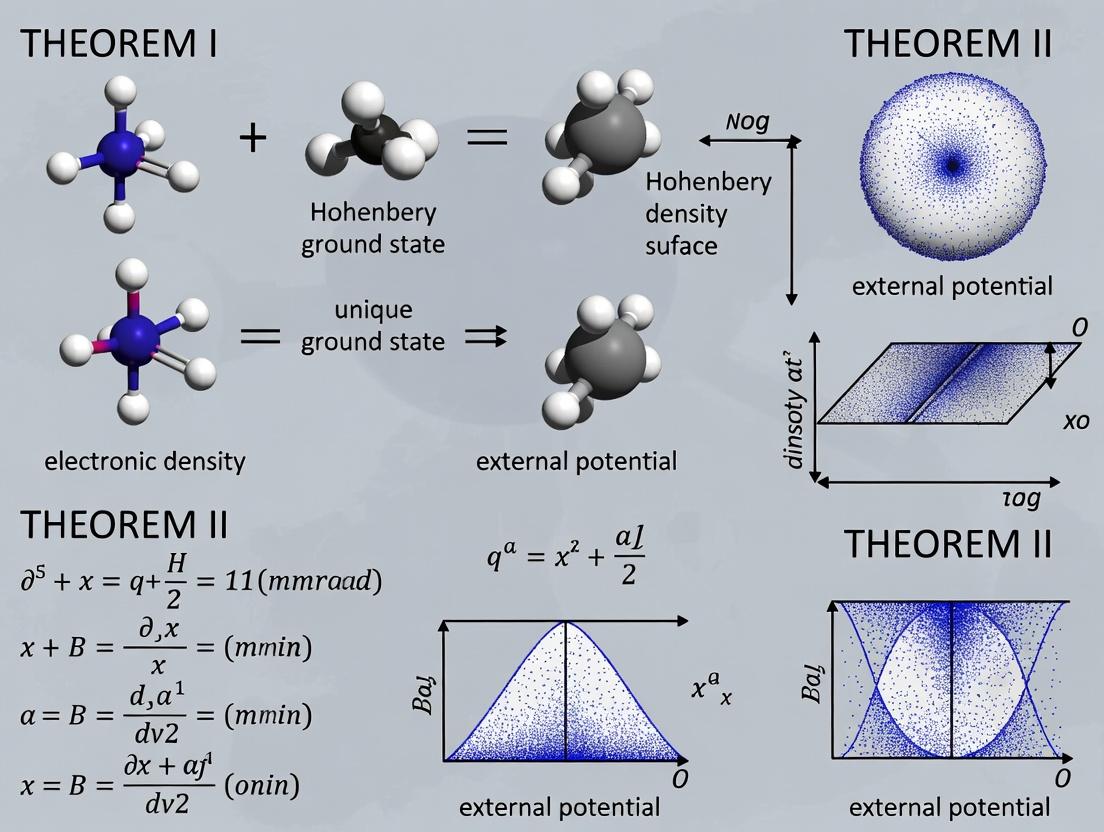
Within the foundational research on Hohenberg-Kohn theorems, the First Theorem establishes the theoretical bedrock for modern density functional theory (DFT). This whitepaper posits that the First HK Theorem is not merely a mathematical curiosity but a profound simplification that redefines the fundamental variables of quantum mechanics for many-electron systems. It shifts the paradigm from the intractable N-electron wavefunction, Ψ(r₁, r₂, ..., r_N), to the observable 3-dimensional electron density, ρ(r). This primacy of ρ(r) provides the direct conceptual link to experimental observables and is the cornerstone upon which all practical DFT calculations in chemistry, materials science, and drug development are built.
Formal Statement and Logical Framework
The First Hohenberg-Kohn Theorem states: The external potential V_ext(r) is (to within an additive constant) a unique functional of the ground-state electron density ρ(r). Since V_ext(r) determines the Hamiltonian (Ĥ), and the Hamiltonian determines all properties of the ground state, it follows that all ground-state properties are unique functionals of the ground-state density.
A critical logical corollary is the one-to-one mapping between key variables. The diagram below illustrates this foundational relationship.
Diagram Title: The HK1 Mapping: Density Determines Potential & All Properties
Methodological & Computational Validation Protocols
The theorem's validity is underpinned by reductio ad absurdum proofs and computational benchmarks. Below is a generalized protocol for validating the theorem's implications via total energy calculations.
Protocol: Benchmarking Energy as a Functional of Density
- System Selection: Choose two distinct molecular systems (e.g., CO and N₂) with identical ground-state electron densities at a single, specific geometry (a mathematical construct, not a physical reality).
- High-Ab Initio Reference:
- Perform a coupled-cluster singles, doubles, and perturbative triples [CCSD(T)] calculation with a large, correlation-consistent basis set (e.g., cc-pVQZ) for each system.
- Extract the accurate ground-state density (ρCCSD(T)(r)) and total energy (Etotal).
- DFT Calculation:
- Use the same density, ρ_CCSD(T)(r), as the input variable for a Kohn-Sham DFT code.
- The code inverts the density to find the corresponding Kohn-Sham potential.
- Analysis: The DFT run would fail to converge to a stable solution or would yield two different external potentials and total energies for the single input density, contradicting the one-to-one mapping. In practice, the theorem is validated by the success of DFT in computing diverse properties from ρ(r).
Quantitative Data: Illustrative Energy Comparisons The following table contrasts the parameter space of wavefunction-based methods versus DFT, highlighting the dimensional reduction mandated by the First HK Theorem.
Table 1: Variable Space Complexity in Electronic Structure Methods
| Method | Fundamental Variable | Spatial Dimensions | Scaling with System Size (N electrons) | Example Computational Cost for N=100 |
|---|---|---|---|---|
| Wavefunction-Based (e.g., CCSD) | Ψ(r₁, r₂, ..., r_N) | 3N | Exponential to ~N⁶-⁷ | Prohibitively large (>10²⁰ ops) |
| Density Functional Theory (DFT) | ρ(r) | 3 | Polynomial ~N³ | Feasible (~10¹⁰ ops) |
Table 2: Validation via Lattice Model Simulations (Numerical Proof)
| Simulation Step | Observable 1 (System A) | Observable 2 (System B) | Conclusion per HK Theorem |
|---|---|---|---|
| Impose identical density ρ(r) | Calculated V_ext(A) | Calculated V_ext(B) | Vext(A) must equal Vext(B) + constant. |
| From derived V_ext | Ground Energy E₀(A) | Ground Energy E₀(B) | E₀(A) and E₀(B) are unique functionals of ρ(r). |
The Scientist's Toolkit: DFT Research Reagents
For researchers employing DFT, whether for catalyst design or ligand-protein binding studies, the following "reagents" are essential.
Table 3: Key Research Reagent Solutions in DFT Calculations
| Reagent / Material | Primary Function | Example in Drug Development Context |
|---|---|---|
| Exchange-Correlation (XC) Functional | Approximates quantum mechanical effects of exchange and correlation; determines accuracy. | B3LYP, PBE: Geometry optimization of drug-like molecules. ωB97X-D, M06-2X: Includes dispersion for protein-ligand binding affinity. |
| Pseudopotential / Basis Set | Represents core electrons and defines the spatial functions for valence electrons. | LANL2DZ: For metal-containing enzymes. def2-TZVP: High-accuracy calculation of partial charges for pharmacophore modeling. |
| Electron Density Grid | Numerical grid for integrating density-dependent properties. | Critical for calculating electrostatic potential maps used in molecular docking. |
| Self-Consistent Field (SCF) Solver | Iteratively solves Kohn-Sham equations to find the ground-state density and energy. | Obtaining a converged electronic structure for transition state analysis of a metabolic reaction. |
| Density of States (DOS) Analyzer | Partitions total energy into orbital contributions. | Analyzing ligand-to-metal charge transfer in photosensitizing drugs. |
Implications and Workflow in Drug Development
The First Theorem enables a practical computational workflow where electron density is the central observable. This workflow connects quantum mechanics to molecular properties relevant to drug design.
Diagram Title: DFT Drug Design Workflow from HK Theorem
The Second Hohenberg-Kohn Theorem, the variational principle for the electron density, is the cornerstone for practical density functional theory (DFT) calculations. It builds upon the First Hohenberg-Kohn Theorem, which establishes a one-to-one mapping between the ground-state electron density ( n(\mathbf{r}) ) and the external potential ( v_{\text{ext}}(\mathbf{r}) ). Within the broader thesis of Hohenberg-Kohn theorems, the second theorem provides the critical actionable principle that transforms DFT from a conceptual framework into a computational workhorse for quantum chemistry and materials science. For researchers and drug development professionals, this theorem enables the prediction of molecular structure, binding energies, and electronic properties essential for rational drug design and materials discovery.
Formal Statement and Mathematical Foundation
The theorem states: For a given external potential ( v_{\text{ext}}(\mathbf{r}) ), the ground-state energy functional ( E_v[n] ) is minimized by the true ground-state electron density ( n_0(\mathbf{r}) ), and the value of the minimum is the true ground-state energy ( E_0 ).
Mathematically, the universal Hohenberg-Kohn energy functional is: [ E{HK}[n] = F{HK}[n] + \int v{\text{ext}}(\mathbf{r}) n(\mathbf{r}) d\mathbf{r} ] where ( F{HK}[n] = T[n] + V{ee}[n] ) is the universal functional (independent of ( v{\text{ext}} )) encompassing the kinetic energy ( T[n] ) and electron-electron interaction ( V{ee}[n] ). The variational principle is: [ E0 = \min{n \in \mathcal{J}N} E{HK}[n] ] where ( \mathcal{J}N ) is the set of ( N )-electron ( v )-representable densities.
Diagram: The HK Theorem Logical Structure
Key Quantitative Data and Functional Approximations
The accuracy of the variational principle hinges on the approximation chosen for ( F_{HK}[n] ). The following table summarizes the evolution and performance of major functional classes.
Table 1: Comparison of Approximate Density Functionals
| Functional Class | Example(s) | Typical Error (kcal/mol) | Computational Cost (Relative) | Key Application in Drug Development |
|---|---|---|---|---|
| Local Density Approximation (LDA) | SVWN5 | 35-50 | 1.0 | Bulk material properties, less used for molecules. |
| Generalized Gradient Approximation (GGA) | PBE, BLYP | 5-15 | 1.1 | Geometry optimization, protein-ligand scaffold screening. |
| Meta-GGA | TPSS, SCAN | 4-10 | 1.5 | Improved binding energies, surface properties. |
| Hybrid GGA | B3LYP, PBE0 | 2-8 | 5-10 | Accurate thermochemistry, reaction barrier prediction. |
| Double Hybrid | B2PLYP, DSD-PBEP86 | 1-4 | 50-100 | High-accuracy benchmark for small molecule drug candidates. |
| Range-Separated Hybrid | ωB97X-D, CAM-B3LYP | 2-6 | 10-15 | Charge-transfer excitations, spectroscopy for chromophores. |
Data compiled from recent benchmark studies (2022-2024). Errors are typical mean absolute deviations for atomization energies and reaction energies.
Experimental and Computational Validation Protocols
The validity of the variational principle is tested by comparing DFT-computed properties against high-level ab initio quantum chemistry methods and experimental data.
Protocol 4.1: Benchmarking Energy Functional Accuracy
Objective: To validate the accuracy of an approximate ( F_{HK}[n] ) for predicting molecular properties.
Materials:
- Benchmark Database: e.g., GMTKN55 (General Main Group Thermochemistry, Kinetics, and Noncovalent interactions) suite.
- Quantum Chemistry Software: Gaussian 16, ORCA, Q-Chem, or CP2K.
- Reference Method: CCSD(T)/CBS (coupled-cluster with perturbative triples, complete basis set limit) data.
Procedure:
- System Selection: Choose a representative subset of molecules and reactions from the benchmark database relevant to drug-like systems (e.g., isomerization energies, barrier heights, non-covalent interaction energies).
- Geometry Optimization: For each species, perform a geometry optimization using the DFT functional under test and a medium-sized basis set (e.g., def2-SVP).
- Single-Point Energy Calculation: Using the optimized geometry, compute a high-accuracy single-point energy with a larger basis set (e.g., def2-QZVP).
- Property Calculation: Compute the target property (e.g., reaction energy ( \Delta E = E{\text{products}} - E{\text{reactants}} )).
- Error Analysis: Calculate the mean absolute error (MAE) and root-mean-square error (RMSE) relative to the reference CCSD(T)/CBS values.
- Statistical Validation: Perform statistical tests to determine if the functional's performance is significantly different from other functionals for the given property set.
Diagram: DFT Benchmarking Workflow
Protocol 4.2: Validating Electron Density via X-ray Diffraction (XRD)
Objective: Experimentally assess the accuracy of the ground-state electron density ( n_0(\mathbf{r}) ) predicted by DFT.
Materials:
- Single Crystal Sample of the target molecule (e.g., a drug candidate).
- High-Resolution X-ray Diffractometer (e.g., synchrotron source).
- Multipolar Model Refinement Software (e.g., MoPro, XD).
- DFT Software for periodic or cluster calculations.
Procedure:
- Data Collection: Collect high-resolution (sin θ/λ > 1.1 Å⁻¹) X-ray diffraction data on a single crystal at low temperature (e.g., 100 K).
- Hansen-Coppens Multipolar Model Refinement: Refine the experimental structure factor data against a multipolar model to obtain the "experimental" electron density ( n_{\text{exp}}(\mathbf{r}) ). This includes modeling deformation density and atomic displacement parameters.
- DFT Calculation: Perform a periodic DFT calculation (or a large-cluster calculation) on the crystal structure using the same geometry. Compute the theoretical electron density ( n_{\text{DFT}}(\mathbf{r}) ).
- Density Difference Analysis: Generate a density difference map: [ \Delta n(\mathbf{r}) = n{\text{exp}}(\mathbf{r}) - n{\text{DFT}}(\mathbf{r}) ]
- Topological Analysis (QTAIM): Perform a quantum theory of atoms in molecules (QTAIM) analysis on both densities to compare critical points (bond critical points, ring critical points), electron densities at these points (( \rho(\mathbf{r}c) )), and Laplacian values (( \nabla^2 \rho(\mathbf{r}c) )).
- Validation: Agreement in topology and small residuals in ( \Delta n(\mathbf{r}) ) maps validate the DFT-predicted density, confirming the practical success of the variational principle in delivering the true ( n_0 ).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Tools for DFT-Based Research
| Item/Reagent | Function/Explanation | Example Vendor/Software |
|---|---|---|
| Exchange-Correlation Functional | Approximates the unknown universal functional ( F_{HK}[n] ); choice dictates accuracy. | Libxc library, built-in functionals in quantum codes. |
| Gaussian-Type Orbital (GTO) Basis Set | Expands the Kohn-Sham orbitals; determines resolution and cost. | def2 series (def2-SVP, def2-TZVP), cc-pVXZ. |
| Plane-Wave/Pseudopotential Set | For periodic solid-state calculations; pseudopotentials replace core electrons. | GBRV, PseudoDojo libraries; PAW datasets (VASP). |
| Implicit Solvation Model | Mimics solvent effects within the DFT framework, crucial for biochemistry. | SMD, COSMO-RS, implemented in packages like Gaussian. |
| Dispersion Correction | Adds empirical van der Waals interactions, essential for non-covalent forces in drug binding. | D3(BJ), D4, MBD corrections. |
| Analysis Software (QTAIM) | Analyzes computed density for bonding insights (bond orders, critical points). | Multiwfn, AIMAll. |
| High-Performance Computing (HPC) Cluster | Provides the necessary computational power for large-scale DFT calculations on proteins or materials. | Local university clusters, cloud HPC (AWS, Azure). |
Advanced Implications: The ( v )-Representability Problem and Levy-Lieb Constrained Search
A critical nuance of the second theorem is the requirement that trial densities be ( v )-representable (i.e., correspond to some antisymmetric wavefunction for some external potential). This condition is difficult to guarantee. The Levy-Lieb constrained search formulation provides a more robust foundation: [ E0 = \min{n} \left{ \min{\Psi \to n} \langle \Psi | \hat{T} + \hat{V}{ee} | \Psi \rangle + \int v_{\text{ext}} n d\mathbf{r} \right} ] This searches first over all wavefunctions yielding a given density ( n ) to define ( F[n] ), then over all ( N )-electron densities. This avoids the ( v )-representability issue by relying on ( N )-representability, which is easier to satisfy.
Diagram: Levy-Lieb Constrained Search Pathway
The Second Hohenberg-Kohn Theorem establishes the indispensable variational principle that makes DFT computationally tractable. For the drug development professional, it underpins every in silico prediction of ligand-protein binding affinity, solvation free energy, and spectroscopic property. While the exact universal functional remains unknown, the continued development of sophisticated approximations, rigorously validated against benchmark protocols, ensures that DFT remains the most widely used electronic structure method across chemistry, materials science, and pharmaceutical research. Its success stands as a testament to the power of the original Hohenberg-Kohn theorems in reducing the many-body wavefunction problem to a more manageable search over three-dimensional densities.
Within the framework of a broader thesis on Hohenberg-Kohn theorems explained research, the v-representability problem emerges as a fundamental, yet often underappreciated, conceptual constraint. The first Hohenberg-Kohn (HK) theorem establishes a one-to-one mapping between the external potential v( r ) (up to an additive constant) and the ground-state electron density n( r ) for a system of interacting electrons. This justifies using the density as the basic variable. However, the practical application of Density Functional Theory (DFT) relies on the second HK theorem, which defines an energy variational principle for the exact density. This is where v-representability becomes critical.
A density is said to be v-representable if it is the ground-state density for some external potential v( r ). The variational principle only guarantees that the exact functional evaluated at the exact ground-state density yields the minimum energy; it does not guarantee that an arbitrary, trial density is v-representable. If a trial density is not v-representable, the functional E[n] may not be defined, or the variational procedure may fail to converge to a physical solution. This problem is circumvented by the Levy-Lieb constrained search formulation, which extends the domain to all N-representable densities (densities derivable from some antisymmetric wavefunction).
Quantitative Data on Functional Performance & Representability
The impact of the v-representability constraint is indirectly observed in the performance and limitations of approximate exchange-correlation functionals. The following table summarizes key benchmarks, highlighting where non-v-representable densities may cause issues.
Table 1: Benchmarking Approximate DFT Functionals and Representability Considerations
| Functional Class | Example(s) | Typical Error (kJ/mol) for Main-Group Thermochemistry | Common Failure Modes Linked to Representability |
|---|---|---|---|
| Local Density Approximation (LDA) | SVWN5 | ~200 | Severe delocalization error, poor for inhomogeneous systems. |
| Generalized Gradient Approximation (GGA) | PBE, BLYP | ~30-50 | Improved but systematic errors for dispersion, band gaps. |
| Meta-GGA | SCAN, TPSS | ~20-30 | Better for diverse bonds, but complexity can lead to numerical instability. |
| Hybrid GGA | B3LYP, PBE0 | ~15-25 | Reduces delocalization error; introduces exact exchange's non-local v-representability. |
| Double Hybrid | B2PLYP, DSD-PBEP86 | ~10-15 | Includes perturbative correlation; computationally intensive. |
| Exact Functional (Theoretical) | N/A | 0 | Defined only for v-representable densities. |
Experimental & Computational Protocols for Addressing Representability
While direct experimental proof of v-representability is impossible, the following computational methodologies are used to probe the limits of DFT approximations, which are implicitly affected by the problem.
Protocol 1: Delocalization Error Assessment via Fractional Charge Calculations
- System Preparation: Construct a series of molecules or atoms with a total number of electrons varied fractionally (e.g., from N-0.5 to N+0.5) using a grand-canonical ensemble approach.
- Energy Calculation: Compute the total energy E(N+δ) as a function of fractional electron count δ using the approximate DFT functional under investigation.
- Analysis: Plot E vs. N. For the exact functional, the curve should be a series of straight line segments. Deviation from linearity (convex curvature) indicates delocalization error, a direct manifestation of the functional's failure for non-integer electron densities, which are non-v-representable.
Protocol 2: Constrained DFT (cDFT) for Studying Charge-Transfer States
- Target State Definition: Define a specific charge or spin state for a subsystem within a larger molecule or complex (e.g., force a metal center to a specific oxidation state).
- Constraint Application: Add a Lagrange multiplier term (w[∫ n( r ) d r - N_target]) to the Kohn-Sham energy functional. This explicitly searches over a subset of densities, bypassing the v-representability issue for the target state.
- Self-Consistent Solution: Solve the modified Kohn-Sham equations iteratively until the constraint is satisfied and the total energy is stationary.
- Validation: Compare results with high-level wavefunction theory (e.g., CASSCF) or experimental spectroscopic data.
Conceptual and Logical Diagrams
Title: Relationship Between Density Classes and the HK Map
Title: Kohn-Sham Self-Consistency Loop
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for DFT Research
| Item / Software | Category | Primary Function in DFT Research |
|---|---|---|
| Gaussian, ORCA, Q-Chem, VASP, CP2K | Electronic Structure Code | Solves the Kohn-Sham equations numerically for molecules or solids, implementing various functionals and algorithms. |
| Pseudo-potentials / PAW Datasets | Core Electron Approximation | Replaces core electrons with an effective potential, drastically reducing computational cost while maintaining accuracy. |
| Basis Sets (e.g., def2-TZVP, cc-pVQZ, plane waves) | Mathematical Basis | Set of functions used to expand Kohn-Sham orbitals; choice balances accuracy and computational expense. |
| LibXC / xcfun Library | Functional Repository | Provides standardized implementations of hundreds of exchange-correlation functionals for code developers. |
| CHEM/BIO Database (e.g., GMTKN55) | Benchmark Database | Collection of chemically relevant benchmark sets to test and validate the accuracy of new DFT functionals. |
| Constraint Implementation (cDFT) | Specialized Algorithm | Allows direct energy minimization over densities constrained to specific properties, sidestepping v-representability for target states. |
This whitepaper details the journey of Density Functional Theory (DFT) from its rigorous mathematical foundations in the Hohenberg-Kohn theorems to its current status as an indispensable tool in materials science, chemistry, and drug development. The broader thesis posits that the Hohenberg-Kohn theorems provided not merely an existence proof but a conceptual scaffold that enabled a sequence of practical approximations, ultimately transforming quantum mechanical simulations from an abstract, numerically intractable problem into a practical computational paradigm. This guide elucidates the core principles, key advancements, and detailed methodologies that define modern DFT.
The Hohenberg-Kohn Theorems: The Foundational Proofs
The 1964 theorems by Pierre Hohenberg and Walter Kohn form the non-negotiable axiomatic base of DFT.
- Theorem I: The ground-state electron density n(r) of a system of interacting electrons in an external potential v(r) (e.g., from nuclei) determines the potential uniquely, up to an additive constant. Since the potential fixes the Hamiltonian, all properties of the ground state are determined by the density.
- Theorem II: A universal energy functional E[n] can be defined for any valid electron density. The exact ground-state density is the one that minimizes this functional, yielding the ground-state energy.
These theorems shift the fundamental variable from the many-body wavefunction Ψ(r₁, r₂, ..., r_N), which depends on 3N coordinates, to the electron density n(*r), a function of only 3 coordinates. This monumental simplification makes realistic calculations on complex systems feasible.
Title: The Logical Flow of the Hohenberg-Kohn Theorems
The Kohn-Sham Equations: The Practical Paradigm
The abstract proofs of Hohenberg and Kohn do not provide a way to compute the energy functional. The practical breakthrough came from Walter Kohn and Lu Jeu Sham in 1965. They introduced a clever mapping of the interacting electron system onto a fictitious system of non-interacting electrons moving in an effective potential, yielding the same ground-state density.
The Kohn-Sham energy functional is partitioned as: EKS[*n*] = *T*s[n] + Eext[*n*] + *E*H[n] + E_xc[n]
Where:
- T_s[n]: Kinetic energy of the non-interacting electrons.
- E_ext[n]: Energy due to external potential.
- E_H[n]: Classical Hartree (electrostatic) energy.
- E_xc[n]: Exchange-Correlation (XC) energy, capturing all remaining quantum mechanical many-body effects.
The variational minimization of EKS[*n*] leads to the Kohn-Sham equations: [ -½∇² + *v*eff(r) ] φi(r) = εi φi(r) with *v*eff(r) = vext(r) + *v*H(r) + vxc(r) and *n*(r) = Σi^N |φ_i(r)|².
These single-particle equations must be solved self-consistently.
Title: The Kohn-Sham Self-Consistent Cycle
The Exchange-Correlation Functional: Accuracy in Practice
The accuracy of a DFT calculation hinges entirely on the approximation chosen for the unknown E_xc[n]. The evolution of these functionals marks the progression of DFT's practical utility.
Table 1: Hierarchy of Common Exchange-Correlation Functionals
| Functional Class | Key Examples | Description | Typical Use Case & Accuracy | ||
|---|---|---|---|---|---|
| Local Density Approximation (LDA) | SVWN | E_xc depends only on the local density n(r). Simple, robust, but overbinds. | Bulk solids, preliminary scans. | ||
| Generalized Gradient Approximation (GGA) | PBE, BLYP | E_xc depends on n(r) and its gradient | ∇n | . Improved bond energies & geometries. | Workhorse for materials & molecules. |
| Meta-GGA | SCAN, TPSS | Adds dependence on kinetic energy density. Better for diverse bonding. | Challenging solids, mixed bonds. | ||
| Hybrid Functionals | B3LYP, PBE0 | Mixes a fraction of exact Hartree-Fock exchange with GGA. Greatly improves molecular properties. | Molecular energetics, band gaps. | ||
| Range-Separated Hybrids | HSE06, ωB97X-D | Separates exchange into short- and long-range parts. Combines accuracy with efficiency for solids. | Accurate band structures, defect levels. |
Table 2: Quantitative Performance of Selected Functionals (Representative Errors)
| Property / System | LDA (SVWN) | GGA (PBE) | Hybrid (HSE06) | Highly Accurate Reference |
|---|---|---|---|---|
| Lattice Constant (Å) | -1 to -2% | ±1% | ±0.5% | Experiment |
| Molecular Bond Energy (kcal/mol) | Error ~30-40 | Error ~5-10 | Error ~2-5 | CCSD(T) |
| Band Gap (eV) - Si | ~0.6 (under) | ~0.6 (under) | ~1.2 (close) | Experimental: 1.17 |
| Reaction Barrier Height | Poor | Moderate | Good | High-level quantum chemistry |
Experimental Protocols: Key Computational Methodologies
Protocol 1: Geometry Optimization and Ground-State Energy
- Initialization: Build atomic structure. Choose functional (e.g., PBE), pseudopotential (e.g., PAW), and plane-wave energy cutoff.
- Self-Consistent Field (SCF): Run the Kohn-Sham cycle (Diagram 2) to convergence (e.g., total energy change < 10⁻⁶ eV).
- Ionic Relaxation: Use forces (calculated via Hellmann-Feynman theorem) to move atoms via conjugate gradient or BFGS algorithm.
- Convergence Check: Repeat SCF and relaxation until forces on all atoms are below a threshold (e.g., 0.01 eV/Å). The final energy is the ground-state energy.
Protocol 2: Electronic Structure Analysis (Band Structure & DOS)
- Ground-State Calculation: Perform a fully converged calculation on a relaxed structure.
- Non-SCF Calculation: Fix the electron density and effective potential from step 1.
- k-Path Generation: Define a high-symmetry path through the Brillouin Zone (e.g., Γ-X-M-Γ).
- Wavefunction Calculation: Solve Kohn-Sham equations for each k-point along the path without updating the density.
- Plotting: Plot eigenvalues ε_n(k) to create the band structure. Compute the Density of States (DOS) by sampling over a dense k-point grid.
Protocol 3: Transition State Search (Nudged Elastic Band - NEB)
- Endpoint Preparation: Fully optimize initial and final state geometries.
- Image Interpolation: Generate 5-10 intermediate "images" along the reaction path.
- NEB Calculation: Run an optimization where each image feels: a) the true atomic forces (projected perpendicular to the path), and b) spring forces (along the path) to maintain even spacing.
- Convergence & Identification: Optimize until images converge. The image with the highest energy is the approximate transition state.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Components of a Modern DFT Simulation
| Item/Component | Function & Explanation |
|---|---|
| Exchange-Correlation Functional | The "reagent" defining the physics. Choices like PBE (general), HSE06 (accurate gaps), or B3LYP (molecules) determine accuracy for a given property. |
| Pseudopotential / PAW Dataset | Replaces core electrons with an effective potential, drastically reducing the number of required plane waves. The "basis set" for plane-wave codes. |
| Plane-Wave Basis Set | Set of periodic functions used to expand Kohn-Sham wavefunctions. Quality controlled by the energy cutoff (E_cut). |
| k-Point Grid | A mesh of points in reciprocal space for Brillouin Zone integration. Finer grids are needed for metals than for insulators. |
| SCF Convergence Criterion | Threshold for stopping the self-consistent cycle (e.g., energy change < 1e-6 Ha). Critical for numerical accuracy. |
| Geometry Convergence Criterion | Threshold for stopping ionic relaxation (e.g., max force < 0.001 Ha/Bohr). Ensures a stable, force-free structure. |
| Dispersion Correction (e.g., D3) | An additive empirical term to account for long-range van der Waals forces, which are missing in most standard functionals. Essential for soft matter, physisorption, and molecular crystals. |
Application in Drug Development: A Practical Frontier
Modern DFT is pivotal in rational drug design, primarily through:
- Enzyme Reaction Mechanism Elucidation: Modeling the catalytic cycle of drug targets (e.g., proteases, kinases) to identify transition states and key interactions.
- Drug-Target Binding Affinity Estimation: Calculating interaction energies between drug candidates and binding pockets, often using hybrid functionals and dispersion corrections.
- Spectroscopic Property Prediction: Simulating NMR chemical shifts, IR vibrational frequencies, and redox potentials to compare with experimental data for lead compound identification.
- Solvation and pKa Modeling: Using implicit (e.g., PCM) or explicit solvation models to understand drug behavior in physiological environments.
Protocol 4: Calculating Ligand-Protein Binding Energy (Simplified)
- Structure Preparation: Isolate a cluster model (80-200 atoms) containing the binding site, cofactors, and ligand from a crystal structure. Saturate dangling bonds with hydrogen atoms.
- Geometry Optimization: Relax the geometry of the protein-ligand complex, the isolated protein site, and the isolated ligand separately using a hybrid functional (e.g., ωB97X-D) and an implicit solvation model.
- Single-Point Energy Calculation: Perform a high-accuracy, well-converged SCF calculation on each optimized structure.
- Energy Analysis: Calculate the interaction energy as ΔE_bind = E(complex) - [E(protein) + E(ligand)]. Apply basis set superposition error (BSSE) correction if using localized basis sets.
The trajectory from the abstract Hohenberg-Kohn theorems to the modern DFT paradigm is a premier example of theoretical physics driving transformative practical innovation. By condensing the many-body problem into an elegant formalism solvable through the Kohn-Sham machinery and progressively more sophisticated exchange-correlation approximations, DFT has become a foundational, high-throughput computational microscope. For researchers and drug development professionals, it offers a quantitative, atomic-scale lens through which to probe electronic structure, reactivity, and interactions, fundamentally accelerating the discovery and design process.
From Theory to Simulation: Implementing Kohn-Sham DFT in Practice
Within the framework of Hohenberg-Kohn (HK) density functional theory (DFT) research, the two foundational theorems establish the existence of a one-to-one mapping between the ground-state electron density ρ(r) and the external potential v_ext(r). While revolutionary, the HK theorems are non-constructive; they confirm the existence of an exact density functional for the total energy, E[ρ], but provide no prescription for its form, especially for the kinetic energy term T[ρ]. The Kohn-Sham (KS) scheme is the crucial bridge that transforms this abstract formalism into a practical, accurate, and widely applicable computational tool.
The Kohn-Sham Ansatz: Reintroducing Orbitals
The central ansatz of Walter Kohn and Lu Jeu Sham (1965) is to replace the intractable, interacting many-electron system with a fictitious system of non-interacting electrons, constrained to have the same ground-state density as the true interacting system.
The exact ground-state density ρ(r) of the interacting system is expressed as a sum over orbitals from the non-interacting system: [ \rho(\mathbf{r}) = \sum{i=1}^{N} |\phii(\mathbf{r})|^2 ] where the ϕ_i are the Kohn-Sham orbitals.
For this non-interacting system, the kinetic energy is known exactly: [ Ts[\rho] = -\frac{1}{2} \sum{i=1}^{N} \langle \phii | \nabla^2 | \phii \rangle ] This Ts is a large and dominant component of the total kinetic energy, and is treated exactly, solving the primary weakness of the original Thomas-Fermi approaches.
The total energy functional is then partitioned as: [ E[\rho] = Ts[\rho] + E{ext}[\rho] + EH[\rho] + E{xc}[\rho] ] Where:
- T_s[ρ]: Non-interacting kinetic energy (exact in KS system).
- Eext[ρ]: Interaction with external potential: ∫ vext(r) ρ(r) dr.
- E_H[ρ]: Classical Hartree (Coulomb) energy: (\frac{1}{2} \iint \frac{\rho(\mathbf{r})\rho(\mathbf{r}')}{|\mathbf{r}-\mathbf{r}'|} d\mathbf{r} d\mathbf{r}').
- Exc[ρ]: Exchange-Correlation (XC) functional. This term encapsulates *everything unknown*: electron correlation, the difference between the true (T) and non-interacting (Ts) kinetic energies, and the non-classical portion of the electron-electron interaction.
The Kohn-Sham Equations
Applying the variational principle to E[ρ] under the constraint that the KS orbitals are orthonormal leads to a set of one-electron Schrödinger-like equations: [ \left[ -\frac{1}{2} \nabla^2 + v{ext}(\mathbf{r}) + vH(\mathbf{r}) + v{xc}(\mathbf{r}) \right] \phii(\mathbf{r}) = \epsiloni \phii(\mathbf{r}) ] These are the Kohn-Sham equations. The effective potential is:
- v_ext(r): External potential (nuclear attraction).
- vH(r): Hartree potential: (vH(\mathbf{r}) = \int \frac{\rho(\mathbf{r}')}{|\mathbf{r}-\mathbf{r}'|} d\mathbf{r}').
- vxc(r): Exchange-correlation potential: (v{xc}(\mathbf{r}) = \frac{\delta E_{xc}[\rho]}{\delta \rho(\mathbf{r})}).
Crucially, these equations must be solved self-consistently, as the potentials depend on the density, which depends on the orbitals.
Title: Self-Consistent Kohn-Sham Cycle.
Key Approximations: The Exchange-Correlation Functional
The entire complexity of the many-body problem is housed within E_xc[ρ]. Its exact form is unknown, and devising accurate approximations is the central challenge in DFT. The following table summarizes the primary hierarchies of functionals.
Table 1: Hierarchy of Common Exchange-Correlation Approximations
| Functional Class | Description | Example(s) | Typical Application & Accuracy | ||
|---|---|---|---|---|---|
| Local Density Approximation (LDA) | E_xc depends only on the local density ρ(r). Exact for a uniform electron gas. | SVWN, PW92 | Solid-state physics (band structures). Tends to overbind molecules. | ||
| Generalized Gradient Approximation (GGA) | E_xc depends on ρ(r) and its gradient | ∇ρ(r) | . Corrects LDA's overbinding. | PBE, BLYP, RPBE | Workhorse for chemistry & materials. Good geometries, decent energies. |
| Meta-GGA | Adds dependence on the kinetic energy density or Laplacian of ρ. | SCAN, TPSS, M06-L | Improved for diverse systems (surfaces, solids, molecules) without Hartree-Fock. | ||
| Hybrid Functionals | Mixes a fraction of exact (Hartree-Fock) exchange with GGA/meta-GGA exchange. | B3LYP, PBE0, HSE06 | Gold standard for molecular thermochemistry, band gaps. More computationally costly. | ||
| Double Hybrids | Adds a perturbative correlation correction on top of hybrid mix. | B2PLYP, DSD-PBEP86 | High-accuracy quantum chemistry, approaching chemical accuracy (±1 kcal/mol). |
Experimental & Computational Protocols in Drug Development
DFT, enabled by the KS scheme, is integral to modern computational drug discovery, providing atomistic insights into electronic structure, binding, and reactivity.
Protocol 1: Calculating Ligand-Protein Binding Affinity (MM/GBSA)
A common endpoint calculation to estimate binding free energies.
Methodology:
- System Preparation: Generate ligand-protein complex, protein alone, and ligand alone structures from molecular dynamics (MD) trajectories or docking.
- Geometry Optimization: Employ DFT (e.g., with PBE or B3LYP functional and a 6-31G* basis set for ligand atoms) to optimize the geometry of snapshots extracted from MD. The protein environment may be treated with a force field.
- Single-Point Energy Calculation: Perform a higher-accuracy DFT calculation (e.g., hybrid functional, larger basis set) on the optimized structures to obtain electronic energies (E_DFT).
- Solvation Energy: Calculate the solvation free energy (ΔG_solv) using an implicit solvation model (e.g., Poisson-Boltzmann/Generalized Born, GB).
- Entropic Contribution: Estimate the conformational entropy change (TΔS) via normal mode analysis or quasi-harmonic approximations on the MD trajectories.
- Free Energy Calculation: Combine terms: [ \Delta G{bind} \approx \Delta E{DFT} + \Delta G{solv} - T\Delta S ] where ΔEDFT = Ecomplex - Eprotein - E_ligand.
Table 2: Research Reagent Solutions for Computational Drug Discovery
| Item/Software | Function in KS-DFT Context | Typical Provider/Implementation |
|---|---|---|
| Quantum Chemistry Code | Solves KS equations, computes energies/properties. | VASP, Gaussian, ORCA, Quantum ESPRESSO, NWChem, CP2K |
| Hybrid/Meta-GGA XC Functional | Provides accurate electronic structure for organic/metal-organic systems. | B3LYP, PBE0, SCAN, ωB97X-D |
| Implicit Solvation Model | Accounts for solvent effects in biological systems. | PCM (Gaussian), SMD (ORCA), VASPsol (VASP) |
| Basis Set Library | Set of mathematical functions to expand KS orbitals. | Pople-type (6-31G*), Dunning-type (cc-pVDZ), Plane waves (with PAW potentials) |
| Pseudopotential/PAW Dataset | Represents core electrons, reduces computational cost for heavy atoms. | GTH (CP2K), US-PP (Quantum ESPRESSO), PAW (VASP) |
Protocol 2: Modeling Reaction Mechanisms in Enzymatic Catalysis
KS-DFT is used to map potential energy surfaces (PES) for biochemical reactions.
Methodology:
- Cluster Model Design: Extract a chemically relevant cluster (200-500 atoms) around the enzyme's active site, saturating dangling bonds with hydrogen atoms. The QM region often includes the substrate, cofactor, and key amino acid residues.
- Geometry Optimization of Stationary Points: Use DFT (e.g., B3LYP-D3/def2-TZVP with an implicit solvation model) to locate reactants (RS), products (PS), and transition states (TS) on the PES.
- Transition State Verification: Confirm the TS structure has a single imaginary vibrational frequency (from Hessian calculation) corresponding to the reaction coordinate motion.
- Intrinsic Reaction Coordinate (IRC) Calculation: Follow the minimum energy path from the TS downhill to confirm it connects the correct RS and PS.
- Energy Refinement: Perform single-point energy calculations on optimized geometries using a higher-level method (e.g., double-hybrid functional or DLPNO-CCSD(T)) and a larger basis set to improve accuracy.
- Energy Decomposition Analysis (EDA): Use specialized DFT methods to dissect interaction energies (e.g., electrostatic, Pauli repulsion, orbital interactions) between fragments (e.g., substrate and enzyme) to understand catalytic contributions.
Title: DFT Modeling of an Enzymatic Reaction Path.
The Kohn-Sham scheme is the indispensable bridge that connects the profound but abstract Hohenberg-Kohn theorems to the vast landscape of practical electronic structure calculations. By cleverly mapping the interacting system onto a tractable non-interacting one, it delegates the major kinetic energy component to exact treatment and isolates the many-body complexity into the exchange-correlation functional. The continuous development of approximate XC functionals, combined with the KS formalism, has made DFT the most widely used method for ab initio calculations in physics, chemistry, and biology. For drug development professionals, it provides a powerful, atomistic toolkit for elucidating ligand-protein interactions, reaction mechanisms, and spectroscopic properties, driving rational design in silico.
The Hohenberg-Kohn (HK) theorems establish the theoretical bedrock of modern density functional theory (DFT). The first theorem proves that the ground-state electron density, n(r), uniquely determines all properties of a many-electron system, including the external potential. The second theorem provides a variational principle: the true ground-state density minimizes the total energy functional E[n]. While monumental, the HK theorems do not provide a practical scheme for calculating this energy or density. The Kohn-Sham (KS) equations, introduced in 1965, solve this by mapping the intractable interacting many-body system onto a fictitious system of non-interacting electrons that yields the same ground-state density. This deconstruction hinges on the precise separation and definition of the kinetic, Hartree, and external potential terms, which is the focus of this technical guide.
Theoretical Deconstruction of the Kohn-Sham Framework
The total energy functional in KS-DFT is decomposed as: [ E{\text{KS}}[n] = Ts[n] + E{\text{ext}}[n] + E{\text{H}}[n] + E_{\text{xc}}[n] ] where:
- ( T_s[n] ): Kinetic energy of the non-interacting Kohn-Sham reference system.
- ( E{\text{ext}}[n] = \int v{\text{ext}}(\mathbf{r}) n(\mathbf{r}) d\mathbf{r} ): Energy due to the external potential (e.g., nuclei).
- ( E_{\text{H}}[n] = \frac{1}{2} \iint \frac{n(\mathbf{r}) n(\mathbf{r}')}{|\mathbf{r}-\mathbf{r}'|} d\mathbf{r} d\mathbf{r}' ): Classical Hartree electron-electron repulsion energy.
- ( E_{\text{xc}}[n] ): Exchange-correlation energy, capturing all remaining quantum mechanical many-body effects.
The variational minimization of ( E{\text{KS}}[n] ) under the constraint of particle conservation leads to the one-electron Kohn-Sham equations: [ \left[ -\frac{1}{2} \nabla^2 + v{\text{eff}}(\mathbf{r}) \right] \psii(\mathbf{r}) = \epsiloni \psii(\mathbf{r}) ] with the effective potential: [ v{\text{eff}}(\mathbf{r}) = v{\text{ext}}(\mathbf{r}) + v{\text{H}}(\mathbf{r}) + v{\text{xc}}(\mathbf{r}) ] and the density constructed from the occupied orbitals: ( n(\mathbf{r}) = \sum{i=1}^{N} |\psi_i(\mathbf{r})|^2 ).
Component Analysis: Kinetic, Hartree, and External Potentials
External Potential ((v_{\text{ext}})): This is the system-defining term, representing the electrostatic attraction between electrons and nuclei, plus any other external fields. It is the only term that differs between systems with different atomic species or geometries.
Kinetic Energy ((Ts[n])): Crucially, ( Ts[n] ) is not the true kinetic energy of the interacting system but that of the auxiliary non-interacting system. The exact, but unknown, kinetic energy of the real system is embedded within the HK functional. The difference is absorbed into the exchange-correlation term: ( T{\text{true}} = Ts + Tc ), where ( Tc ) is the correlation kinetic energy, part of ( E_{\text{xc}} ).
Hartree Potential ((v{\text{H}})): Derived from ( \delta E{\text{H}} / \delta n ), it is the classical mean-field repulsion from the total electron density: ( v_{\text{H}}(\mathbf{r}) = \int \frac{n(\mathbf{r}')}{|\mathbf{r}-\mathbf{r}'|} d\mathbf{r}' ). It includes an unphysical self-interaction for each electron, which a functional exchange term must cancel.
Exchange-Correlation Potential ((v{\text{xc}})): The functional derivative ( v{\text{xc}}(\mathbf{r}) = \delta E{\text{xc}}[n] / \delta n(\mathbf{r}) ) is the most critical and challenging term, encompassing all quantum mechanical effects: exchange, correlation, the kinetic energy difference ((Tc)), and self-interaction correction.
Quantitative Comparison of DFT Potentials and Functionals
The accuracy of a KS-DFT calculation is determined almost entirely by the approximation chosen for ( E_{\text{xc}}[n] ). The following table summarizes key classes of functionals and their handling of the core potentials.
Table 1: Hierarchy of Exchange-Correlation Functionals and Their Characteristics
| Functional Class | Examples | Treatment of (E_{\text{xc}}) | (T_s) Handling | Self-Interaction Error | Typical Application |
|---|---|---|---|---|---|
| Local Density Approx. (LDA) | SVWN | (E{\text{xc}} = \int n(\mathbf{r}) \epsilon{\text{xc}}^{\text{hom}}(n(\mathbf{r})) d\mathbf{r}) | Exact for KS system | High | Bulk solids, uniform electron gas |
| Generalized Gradient Approx. (GGA) | PBE, BLYP | (E_{\text{xc}} = \int f(n(\mathbf{r}), \nabla n(\mathbf{r})) d\mathbf{r}) | Exact for KS system | Moderate | General-purpose chemistry, materials |
| Meta-GGA | SCAN, TPSS | (E_{\text{xc}} = \int g(n, \nabla n, \tau) d\mathbf{r}), (\tau) is kinetic energy density | Implicitly uses (\tau) from KS orbitals | Lower | Diverse systems, improved bonds |
| Hybrid | B3LYP, PBE0 | Mixes exact (Hartree-Fock) exchange with DFT exchange-correlation: (a Ex^{\text{HF}} + (1-a)Ex^{\text{DFT}} + E_c^{\text{DFT}}) | Exact HF exchange uses occupied orbitals | Reduced | Molecular thermochemistry, band gaps |
| Double Hybrid | B2PLYP | Adds MP2-like correlation: Mixes exact exchange with DFT exchange, plus perturbative correlation | Uses both occupied and virtual orbitals | Very Low | High-accuracy quantum chemistry |
Table 2: Magnitude Comparison of Energy Components for a Representative Molecule (H₂O, PBE/def2-TZVPP Level)
| Energy Component | Value (Hartree) | % of Total Energy | Notes | ||
|---|---|---|---|---|---|
| Total Energy (E_{\text{KS}}) | -76.438 | 100% | Converged SCF result | ||
| Kinetic Energy (T_s) | 76.120 | ~99.6% (of ( | E | )) | Large and positive |
| External Potential Energy (E_{\text{ext}}) | -200.547 | 262% | Large and negative, dominant attractive term | ||
| Hartree Energy (E_{\text{H}}) | 46.672 | 61% | Positive repulsive term | ||
| XC Energy (E_{\text{xc}}) | -9.193 | 12% | Moderately sized corrective term |
Experimental & Computational Protocols
This section outlines a standard protocol for performing a KS-DFT calculation, emphasizing the role of the deconstructed potentials.
Protocol 1: Standard Self-Consistent Field (SCF) Cycle for Solving Kohn-Sham Equations
Objective: To obtain the ground-state electron density and total energy for a given atomic structure and XC functional.
Materials (Computational):
- Initial Atomic Coordinates & Basis Set: A molecular geometry file (e.g., .xyz, .cif) and a predefined set of basis functions (e.g., Gaussian-type orbitals, plane waves).
- XC Functional: The mathematical approximation for (E_{\text{xc}}[n]) (e.g., PBE).
- KS-DFT Software: Package such as Gaussian, ORCA, VASP, Quantum ESPRESSO, or PySCF.
Methodology:
- Initialization: Construct the initial guess for the electron density (n^{(0)}(\mathbf{r})). Common methods include superposition of atomic densities or a simple Hückel guess.
- Build Effective Potential: For iteration (k): a. Compute the external potential (v{\text{ext}}) from nuclear charges and positions. b. Compute the Hartree potential (v{\text{H}}^{(k)}) by solving the Poisson equation for the current density (n^{(k)}). c. Compute the XC potential (v{\text{xc}}^{(k)}) as the functional derivative of the chosen (E{\text{xc}}) at density (n^{(k)}). d. Sum components: (v{\text{eff}}^{(k)} = v{\text{ext}} + v{\text{H}}^{(k)} + v{\text{xc}}^{(k)}).
- Solve Kohn-Sham Equations: Insert (v{\text{eff}}^{(k)}) into the one-electron KS Hamiltonian and diagonalize it within the chosen basis set to obtain a new set of orbitals ({\psii^{(k+1)}}) and eigenvalues ({\epsilon_i^{(k+1)}}).
- Form New Density: Construct the new density from occupied orbitals: (n^{(k+1)}(\mathbf{r}) = \sum{i}^{\text{occ}} |\psii^{(k+1)}(\mathbf{r})|^2).
- Check Convergence: Assess if the change in density ((||n^{(k+1)} - n^{(k)}||)) and/or total energy ((|E^{(k+1)} - E^{(k)}|)) is below a predefined threshold (e.g., (10^{-6}) Ha). If not, use a density mixing scheme (e.g., Pulay, Kerker) to generate an input density for the next iteration and return to Step 2.
- Post-Processing: Upon convergence, use the final density, orbitals, and energies to compute derived properties: forces, vibrational frequencies, electronic spectra, etc.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key "Research Reagent Solutions" in Computational KS-DFT Studies
| Item | Category | Function & Relevance to KS Potentials |
|---|---|---|
| Pseudopotentials / PAWs | Core Potential Replacement | Replace (v_{\text{ext}}) from core electrons with an effective potential, drastically reducing the number of explicit electrons to be calculated. Critical for heavy elements. |
| Gaussian Basis Sets | Orbital Representation (Chemistry) | Pre-defined sets of functions (e.g., cc-pVTZ, def2-TZVP) used to expand the KS orbitals (\psi_i(\mathbf{r})). Accuracy defines the completeness of the Hilbert space. |
| Plane-Wave Basis Sets | Orbital Representation (Materials) | Expands KS orbitals in Fourier space. Quality controlled by a kinetic energy cut-off. Naturally periodic, ideal for solids and materials. |
| XC Functional Library | Physical Model | The defining "reagent" determining accuracy. Libraries like Libxc provide hundreds of tested (E{\text{xc}}[n]) and (v{\text{xc}}[n]) implementations. |
| Density Mixing Algorithms | Convergence Aid | Algorithms (e.g., DIIS, Kerker) that stabilize the SCF cycle by intelligently mixing densities from previous iterations to construct (n^{(k)}_{\text{input}}), preventing charge sloshing. |
| Linear-Scaling Solvers | Computational Engine | Algorithms (e.g., Conquest, ONETEP) that exploit locality to solve KS equations with computational cost scaling linearly with system size, enabling large-scale simulations. |
Visualizing the Kohn-Sham Framework and Workflow
Diagram 1: Kohn-Sham Self-Consistent Field Cycle
Diagram 2: From Hohenberg-Kohn Theorems to KS Applications
Within the rigorous framework established by the Hohenberg-Kohn theorems, which prove that the ground-state electron density uniquely determines all properties of a many-electron system, Density Functional Theory (DFT) has become the cornerstone of computational quantum chemistry and materials science. The theorems, however, do not specify the form of the universal functional ( F[\rho] ), which contains the kinetic energy of non-interacting electrons and the electron-electron interaction energy. The critical, and famously unknown, component of this functional is the exchange-correlation (XC) energy, ( E{XC}[\rho] ), which encapsulates all quantum mechanical many-body effects. The accuracy of any DFT calculation hinges entirely on the approximation used for ( E{XC} ). This guide provides an in-depth analysis of the primary families of XC functionals, their evolution, and their application in modern research, particularly for drug development professionals investigating molecular interactions, binding affinities, and electronic properties of bioactive compounds.
The Hierarchy of Exchange-Correlation Functionals
Local Density Approximation (LDA)
LDA is the simplest approximation, derived from the homogeneous electron gas (HEG) model. It assumes the XC energy density at a point in space depends only on the electron density at that point. [ E{XC}^{LDA}[\rho] = \int \rho(\mathbf{r}) \, \epsilon{xc}^{HEG}(\rho(\mathbf{r})) \, d\mathbf{r} ] While LDA provides robust structures and is computationally efficient, it suffers from systematic errors, including overbinding and poor description of molecular dissociation energies.
Generalized Gradient Approximation (GGA)
GGA improves upon LDA by including the gradient of the density ( \nabla\rho(\mathbf{r}) ), accounting for inhomogeneity. [ E{XC}^{GGA}[\rho] = \int \rho(\mathbf{r}) \, \epsilon{xc}^{GGA}(\rho(\mathbf{r}), \nabla\rho(\mathbf{r})) \, d\mathbf{r} ] GGAs are separated into exchange and correlation parts, developed semi-empirically or via constraints. They correct LDA's overbinding and improve bond energies and geometries.
Meta-GGA
Meta-GGAs incorporate additional ingredients beyond the density and its gradient, typically the kinetic energy density ( \tau(\mathbf{r}) ) or the Laplacian of the density ( \nabla^2\rho(\mathbf{r}) ). [ E{XC}^{Meta-GGA}[\rho] = \int \rho(\mathbf{r}) \, \epsilon{xc}(\rho(\mathbf{r}), \nabla\rho(\mathbf{r}), \tau(\mathbf{r})) \, d\mathbf{r} ] This allows them to satisfy more exact constraints and improve accuracy for diverse properties, including transition states and solid-state properties, without a significant computational leap from GGAs.
Hybrid Functionals
Hybrid functionals mix a portion of exact Hartree-Fock (HF) exchange with DFT exchange-correlation, based on the adiabatic connection theorem. The general form is: [ E{XC}^{Hybrid} = a EX^{HF} + (1-a) EX^{DFT} + EC^{DFT} ] System-specific (e.g., HSE) or range-separated hybrids (e.g., ωB97X-D) are crucial for accurately predicting band gaps, reaction barriers, and non-covalent interactions critical in drug design.
Quantitative Comparison of XC Functional Performance
Table 1: Benchmark Performance of Common XC Functionals on Key Molecular Properties.
| Functional Class | Example | Atomization Energy Error (kcal/mol) | Band Gap Error (eV) | Non-Covalent Interaction Error | Computational Cost Factor |
|---|---|---|---|---|---|
| LDA | SVWN5 | ~30-40 | Severe Underestimation | Poor | 1.0 |
| GGA | PBE | ~10-15 | Moderate Underestimation | Fair | ~1.1 |
| Meta-GGA | SCAN | ~4-6 | Improved but Underestimated | Good | ~1.5 |
| Hybrid | PBE0 | ~3-5 | Improved but Underestimated | Good | ~10-100 |
| Range-Separated Hybrid | ωB97X-V | ~1-2 | Good Accuracy | Excellent | ~50-200 |
Table 2: Suitability for Drug Development Applications.
| Application | Recommended Functional(s) | Key Rationale |
|---|---|---|
| Protein-Ligand Binding Energy | ωB97X-D, DSD-PBEP86-D3(BJ) | Accurate treatment of dispersion and charge transfer. |
| Reaction Mechanism (Enzyme) | M06-2X, ωB97X-D | Good for transition states and barrier heights. |
| Molecular Geometry & Vibrations | PBE0, B3LYP-D3(BJ) | Reliable structures and frequencies at moderate cost. |
| Electronic Excitation (UV-Vis) | CAM-B3LYP, ωB97X-D | Correct long-range behavior for charge-transfer states. |
| Solvation Free Energy | M06-2X, SMD(ωB97X-D) | Coupling with implicit solvation models. |
Experimental Protocols for Benchmarking XC Functionals
Protocol 1: Benchmarking Binding Affinity (ΔG) for a Protein-Ligand Complex
- System Preparation: Obtain 3D structures from PDB. Add hydrogens, assign protonation states (e.g., using H++ or PROPKA at pH 7.4).
- Geometry Optimization: Optimize ligand and protein-binding site (or full protein) using a medium-tier hybrid functional (e.g., PBE0) and a double-zeta basis set with dispersion correction (e.g., def2-SVP with D3(BJ)).
- Single-Point Energy Calculation: Perform a high-accuracy single-point calculation on the optimized geometry using a robust, dispersion-corrected hybrid or double-hybrid functional (e.g., DLPNO-CCSD(T)/CBS or ωB97X-V/def2-QZVPP) as a reference. Repeat with the functionals being tested (e.g., a GGA, meta-GGA, and hybrid).
- Binding Energy Calculation: Compute the interaction energy as ( E{complex} - (E{protein} + E_{ligand}) ). Apply Basis Set Superposition Error (BSSE) correction via the Counterpoise method.
- Validation: Compare computed ΔG values against experimentally measured binding constants (e.g., from ITC or SPR). Statistical analysis (MAE, RMSE) determines functional accuracy.
Protocol 2: Assessing Electronic Excitation Energies for a Chromophore
- Ground-State Optimization: Fully optimize the chromophore's geometry using the target functional and a polarized basis set.
- Excitation Calculation: Perform Time-Dependent DFT (TD-DFT) calculations on the optimized structure using the same and other XC functionals (especially range-separated hybrids).
- Analysis: Extract the lowest 10-20 vertical excitation energies and oscillator strengths.
- Benchmarking: Compare the computed lowest singlet excitation energy (S0→S1) against the experimental maximum absorption wavelength (λ_max) from UV-Vis spectroscopy. Higher-level wavefunction methods (e.g., EOM-CCSD) can serve as computational benchmarks.
Computational Workflow for DFT-Based Drug Discovery
Title: DFT Drug Discovery Workflow.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Computational Tools for XC Functional Research & Application.
| Item / Software | Function / Purpose | Key Utility |
|---|---|---|
| Quantum Chemistry Packages (Gaussian, ORCA, Q-Chem, NWChem) | Perform SCF, geometry optimization, frequency, TD-DFT, and post-HF calculations. | Implementation of hundreds of XC functionals; essential for all protocols. |
| Dispersion Correction (D3, D3(BJ), VV10) | Add empirical van der Waals corrections to DFT energies. | Crucial for obtaining accurate non-covalent interaction energies in drug binding. |
| Implicit Solvation Models (SMD, COSMO) | Model solvent effects without explicit solvent molecules. | Provides realistic solvation free energies and shifts in electronic properties. |
| Basis Sets (def2-SVP, def2-TZVP, cc-pVDZ, 6-31G*) | Sets of mathematical functions representing atomic orbitals. | Balance between accuracy and computational cost; essential for any calculation. |
| Wavefunction Analysis (Multiwfn, AIMAll) | Analyze electron density, orbitals, electrostatic potential (ESP). | Visualizes bonding, charges, and reactivity patterns predicted by the XC functional. |
| Benchmark Databases (GMTKN55, S66, NCIE) | Curated sets of molecules with reference energies (experimental/high-level theory). | Gold standard for validating and ranking the accuracy of new and existing XC functionals. |
Logical Map of XC Functional Development
Title: Evolution Path of XC Functionals.
The journey from the foundational Hohenberg-Kohn theorems to practical drug discovery simulations is paved by successive approximations for the exchange-correlation functional. The choice of functional—from efficient GGAs for preliminary screening to sophisticated, system-tuned hybrids for final prediction—directly dictates the reliability of computed properties. For the drug development researcher, this translates to a strategic balance between computational cost and the required accuracy for binding energies, reaction pathways, or spectroscopic predictions. As the field evolves towards incorporating machine learning and higher-level constraints, the core principle remains: a deep understanding of the strengths and pathologies of each XC functional family is essential for generating chemically meaningful and predictive results.
This technical guide explores the application of Density Functional Theory (DFT) within modern computational drug discovery. Framed by the foundational Hohenberg-Kohn theorems—which establish the one-to-one mapping between a system's ground-state electron density and its external potential—DFT provides a practical quantum mechanical framework for predicting molecular properties critical to pharmaceutical development.
Theoretical Foundation and Protocol
The Hohenberg-Kohn theorems justify the use of electron density as the fundamental variable, reducing the many-body Schrödinger equation problem to tractable calculations for large biomolecular systems. The standard workflow for assessing ligand-protein binding involves multiple DFT-based steps.
Diagram: DFT in Drug Design Workflow
Key Computational Experiments and Data
1. Binding Affinity via QM/MM: A hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) protocol is employed, where the ligand and key protein residues are treated with DFT, while the remainder is handled with molecular mechanics.
Experimental Protocol:
- System Preparation: A protein-ligand complex is extracted from a crystal structure (e.g., PDB ID). The ligand and residues within 5Å are designated the QM region (typically treated with B3LYP/6-31G(d)). The system is solvated in an explicit water box.
- Geometry Optimization: The QM region is optimized using DFT, with the MM region fixed or restrained.
- Single-Point Energy Calculation: A high-level single-point energy calculation (e.g., ωB97XD/def2-TZVP) is performed on the optimized QM region.
- Binding Energy Calculation: The interaction energy is computed as ΔE_bind = E(complex) - [E(protein) + E(ligand)], often with corrections for basis set superposition error (BSSE).
2. Reactivity Descriptor Analysis: Global and local reactivity indices, grounded in Conceptual DFT, are calculated from DFT-derived energies and electron density.
Experimental Protocol:
- Single Molecule Calculations: The ligand is optimized using a functional like B3LYP and basis set like 6-311++G(d,p).
- Energy Calculations: Single-point calculations are performed on the Neutral (N), Cation (N+1), and Anion (N-1) species.
- Descriptor Computation:
- Chemical Potential (μ) ≈ (EHOMO + ELUMO)/2
- Hardness (η) ≈ (ELUMO - EHOMO)
- Fukui Functions (f⁺, f⁻) are computed via finite difference analysis of electron densities to identify nucleophilic/electrophilic sites.
Table 1: DFT-Calculated Reactivity Descriptors for Protease Inhibitors
| Ligand ID | Target (PDB) | E_HOMO (eV) | E_LUMO (eV) | Chemical Potential, μ (eV) | Global Hardness, η (eV) | Predicted ΔG_bind (QM/MM, kcal/mol) | Experimental IC₅₀ (nM) |
|---|---|---|---|---|---|---|---|
| LIG-01 | 7TLL | -6.21 | -1.85 | -4.03 | 2.18 | -9.7 | 12.5 |
| LIG-02 | 7TLL | -5.89 | -1.92 | -3.91 | 1.99 | -11.2 | 3.8 |
| LIG-03 | 6WUF | -7.05 | -0.98 | -4.02 | 3.04 | -8.1 | 110.0 |
Table 2: Key Research Reagent Solutions & Computational Tools
| Item Name | Category | Function in DFT Drug Design |
|---|---|---|
| B3LYP Functional | DFT Method | Hybrid functional offering balance of accuracy & cost for geometry optimization. |
| def2-TZVP Basis Set | Basis Set | Triple-zeta basis for high-accuracy single-point energy & property calculations. |
| CPCM/SMD Solvation Model | Implicit Solvent | Models bulk solvent effects (e.g., water) critical for biological simulations. |
| Quantum Espresso/Gaussian | Software Suite | Platforms for performing DFT energy, optimization, and wavefunction analysis. |
| PDB Fixer (OpenMM) | Preprocessing Tool | Prepares & fixes protein structures from the PDB for QM/MM simulations. |
| Multiwfn/VMD | Analysis Tool | Analyzes electron density, computes Fukui functions, and visualizes results. |
Pathway and Interaction Analysis
DFT elucidates interaction mechanisms at the electronic level. For a kinase inhibitor, the bonding can be decomposed.
Diagram: DFT-Electronic Interactions in Binding
In conclusion, anchored by the rigorous framework of the Hohenberg-Kohn theorems, DFT transitions from abstract quantum theory to an indispensable tool in drug design. By providing quantitative predictions of binding affinities and revealing atomic-scale reactivity, it directly informs the rational design of novel therapeutic agents.
This whitepaper examines the practical applications of Density Functional Theory (DFT) within materials science, framed by the foundational Hohenberg-Kohn theorems. The first theorem establishes the one-to-one mapping between an external potential and the ground-state electron density, while the second provides the variational principle for the energy functional. These theorems legitimize the use of electron density as the fundamental variable, enabling the computational exploration of complex materials properties. For researchers and drug development professionals, DFT serves as a critical in silico tool for predicting electronic structure, catalytic activity, and interfacial interactions, guiding experimental synthesis and characterization.
Core Applications and Methodologies
Band Gap Calculations
The accurate prediction of electronic band gaps is crucial for semiconductor and photovoltaic material design. DFT calculations, while efficient, are known to underestimate band gaps due to approximations in the exchange-correlation functional.
Experimental Protocol: Band Structure Calculation
- Structure Optimization: Obtain the crystal structure from databases (e.g., ICSD, COD) or experimental refinement. Perform geometry optimization until forces on all atoms are below 0.01 eV/Å and stresses are minimized.
- Self-Consistent Field (SCF) Calculation: Perform a converged SCF calculation on the optimized structure using a plane-wave basis set and pseudopotentials. A k-point mesh density of at least 0.03 1/Å is recommended.
- Band Structure Unfolding: Execute a non-self-consistent field (NSCF) calculation along high-symmetry paths in the Brillouin zone (e.g., Γ-X-M-Γ for cubic systems). Extract eigenvalues to plot the electronic band structure.
- Band Gap Extraction: Identify the valence band maximum (VBM) and conduction band minimum (CBM). The fundamental band gap is calculated as Egap = ECBM - E_VBM. For more accurate results, hybrid functionals (e.g., HSE06) or GW corrections are employed post-GGA.
Title: DFT Workflow for Band Gap Calculation
Table 1: DFT-Calculated vs. Experimental Band Gaps for Selected Semiconductors
| Material | DFT Functional | Calculated Gap (eV) | Experimental Gap (eV) | % Error |
|---|---|---|---|---|
| Silicon | PBE | 0.6 | 1.17 | -48.7% |
| Silicon | HSE06 | 1.2 | 1.17 | +2.6% |
| TiO₂ (Anatase) | PBE | 2.1 | 3.2 | -34.4% |
| TiO₂ (Anatase) | HSE06 | 3.3 | 3.2 | +3.1% |
| GaN | PBE | 1.7 | 3.4 | -50.0% |
| GaN | GW | 3.5 | 3.4 | +2.9% |
Catalytic Activity Prediction
DFT enables the calculation of reaction pathways on catalytic surfaces. Key metrics include adsorption energies, reaction energies, and activation barriers, which correlate with catalytic activity and selectivity.
Experimental Protocol: Adsorption Energy and Reaction Pathway
- Surface Slab Construction: Create a periodic slab model of the catalyst surface (e.g., (111) facet for fcc metals) with sufficient vacuum (≥ 15 Å) to avoid periodic interactions.
- Adsorbate Placement: Place the reactant molecule(s) at plausible adsorption sites (e.g., atop, bridge, hollow) on the relaxed surface slab.
- Transition State Search: Use methods like the Nudged Elastic Band (NEB) or Dimer method to locate the saddle point (transition state) between initial and final states. Confirm with frequency analysis (one imaginary frequency).
- Energy Calculation: Calculate the adsorption energy: Eads = E(surface+adsorbate) - Esurface - Eadsorbate. Compute the reaction energy (ΔE) and activation barrier (E_a) from the located intermediates and transition states.
Title: DFT Pathway for Catalytic Surface Reaction
Table 2: DFT-Calculated Catalytic Parameters for CO Oxidation on Pt(111)
| Reaction Step | Calculated Energy (eV) | Key Parameter |
|---|---|---|
| CO adsorption | -1.45 | Adsorption Energy |
| O₂ adsorption & dissociation | -0.98 (O₂) / 0.21 (Barrier) | Dissociation Barrier |
| CO + O → CO₂ (via TS) | -3.12 (Reaction) / 0.87 (Barrier) | Activation Energy (E_a) |
Surface Interaction Analysis
Understanding molecular adsorption on material surfaces is vital for sensor design, corrosion prevention, and drug delivery systems (e.g., functionalized nanoparticles). DFT provides insights into binding modes, charge transfer, and electronic structure modifications.
Experimental Protocol: Surface Adsorption and Charge Analysis
- Model System Preparation: Build a surface model (slab) and the adsorbate molecule. For biomolecules, consider relevant protonation states at physiological pH.
- Binding Site Screening: Systematically test all symmetry-inequivalent adsorption sites. Perform constrained optimizations for each configuration.
- Electronic Analysis: Perform a single-point calculation on the lowest-energy adsorption structure. Conduct a Bader charge or Mulliken population analysis to quantify charge transfer. Calculate the projected density of states (PDOS) to identify orbital interactions.
- Binding Affinity: The binding energy is computed as: Ebind = E(slab+mol) - Eslab - Emol. A more negative value indicates stronger binding.
The Scientist's Toolkit: Research Reagent Solutions
| Item/Reagent | Function in DFT-Guided Research |
|---|---|
| VASP, Quantum ESPRESSO, CASTEP | Software packages for performing DFT calculations with plane-wave basis sets and pseudopotentials. |
| PBE, PW91, RPBE Functionals | Generalized Gradient Approximation (GGA) exchange-correlation functionals for standard geometry and energy calculations. |
| HSE06, B3LYP Hybrid Functionals | More accurate (but costly) functionals incorporating exact Hartree-Fock exchange for improved band gaps and reaction energies. |
| Projector Augmented-Wave (PAW) Potentials | Pseudopotentials that accurately describe valence electron interactions while treating core electrons efficiently. |
| VESTA, Jmol | Visualization software for constructing crystal structures, surfaces, and plotting charge density isosurfaces. |
| NEB (Nudged Elastic Band) Code | Algorithm for finding minimum energy paths and transition states between known reactant and product states. |
| Materials Project, C2DB Databases | Online repositories of pre-computed DFT data for thousands of materials, used for validation and benchmarking. |
Navigating DFT Calculations: Accuracy Limits, Common Pitfalls, and Best Practices
The Hohenberg-Kohn (HK) theorems establish the existence of a unique energy functional, E[ρ], whose minimum yields the exact ground-state electron density and energy. However, the exact form of the universal functional—specifically the exchange-correlation (XC) part, EXC[ρ]—is unknown. Practical Density Functional Theory (DFT) relies on approximations to EXC (e.g., LDA, GGA, hybrid functionals). This work examines two pervasive and interrelated failures of these approximations that stem directly from the inexact description of E_XC: the Self-Interaction Error (SIE) and the associated problem of excessive electron delocalization.
Theoretical Foundations and Quantitative Manifestations
2.1 The Self-Interaction Error (SIE) In exact DFT, the Hartree energy of an electron interacting with itself is perfectly canceled by the exchange term. Approximate functionals lack this full cancellation, leading to SIE. This error systematically stabilizes delocalized electron densities over localized ones.
Table 1: Quantitative Impact of SIE on Reaction Barrier Heights and Band Gaps
| System/Property | Exact/Exp. Value | LDA Result | GGA (PBE) Result | Hybrid (PBE0) Result | Notes |
|---|---|---|---|---|---|
| H₂⁺ Dissociation Energy (eV) | Exact: -4.58 eV | ~-5.1 eV | ~-4.9 eV | ~-4.6 eV | SIE severely affects one-electron systems. |
| H + H₂ → H₂ + H Barrier (eV) | Exp.: ~0.4 eV | ~0.1 eV | ~0.2 eV | ~0.3 eV | SIE underestimates barriers, favoring charge delocalization in transition state. |
| Si Band Gap (eV) | Exp.: 1.17 eV | ~0.5 eV | ~0.6 eV | ~1.2 eV | SIE causes severe underestimation of band gaps in semiconductors (charge delocalization over lattice). |
| Charge Transfer Excitation in Zn²⁺-Porphyrin–Quinone (eV) | TD-DFT Benchmark: ~2.8 eV | N/A | ~1.5 eV | ~2.5 eV | SIE in GGA drastically underestimates CT state energy. |
2.2 The Delocalization Problem SIE inherently biases electron densities toward excessive delocalization. This manifests in incorrect predictions for molecular dissociation limits, charge-localized states in transition-metal complexes, and charge-transfer excitations.
Experimental and Computational Methodologies for Diagnosis
3.1 Protocol: Diagnosing SIE via the DFT+U Method (for Transition Metal Oxides)
- System Preparation: Construct a supercell of the transition metal oxide (e.g., NiO, a charge-transfer insulator).
- Baseline Calculation: Perform a standard spin-polarized DFT calculation using a GGA functional (e.g., PBE) on the supercell. Analyze the electronic density of states (DOS) and projected DOS (pDOS) onto metal d-orbitals.
- DFT+U Application: Introduce an on-site Coulomb repulsion parameter, U, and an exchange parameter, J (often as U_eff = U - J), to the metal d-orbitals. Typical values for NiO: U_eff ≈ 6-8 eV.
- Calculation & Comparison: Recalculate the electronic structure with the same basis set and k-point mesh. Compare the band gap, magnetic moment, and pDOS with the baseline GGA results and experimental data.
- Analysis: The GGA result will show metallic or small-gap behavior with overly delocalized d-electrons. DFT+U corrects this by imposing an energy penalty on delocalization, opening a band gap and localizing electrons, better matching the antiferromagnetic insulating state observed experimentally.
3.2 Protocol: Evaluating Delocalization in Organic Radicals
- Target Systems: Select organic radicals where localization is key (e.g., benzyl radical, phenoxyl radical).
- Charge Population Analysis: Perform geometry optimization with a suite of functionals (LDA, GGA, global hybrid, range-separated hybrid). Use population analysis schemes (e.g., Hirshfeld, Mulliken, or Löwdin) to assess the spin density distribution.
- Comparison Metric: Quantify the deviation from idealized integer spin population on the radical center. Calculate the magnitude of spurious spin density on adjacent non-radical atoms.
- Benchmarking: Compare against high-level wavefunction theory (e.g., CCSD(T)) or experimental hyperfine coupling constants. Functionals with lower SIE (e.g., range-separated hybrids) will show spin densities more localized on the radical center.
Visualizing the Relationship between SIE, Delocalization, and Functional Approximations
Title: Causal Pathway from DFT Approximations to Physical Errors
Title: Functional Development Path to Mitigate SIE and Delocalization
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Tools and Functionals for SIE/Delocalization Research
| Item/Reagent (Computational Functional/Method) | Primary Function & Role in Addressing SIE/Delocalization |
|---|---|
| Local Density Approximation (LDA) | Baseline functional; exhibits severe SIE, leading to maximal delocalization. Used as a control for error magnitude. |
| Generalized Gradient Approximation (GGA) | Improves upon LDA but retains significant SIE. Workhorse for geometry optimization but unreliable for properties sensitive to electron localization. |
| Global Hybrid Functional (e.g., B3LYP, PBE0) | Mixes a fraction (20-25%) of exact Hartree-Fock exchange with GGA. Partially cancels SIE, improving band gaps and barrier heights. Standard for molecular chemistry. |
| Range-Separated Hybrid Functional (e.g., CAM-B3LYP, ωB97X-D) | Treats exact exchange differently at short- and long-range. Dramatically improves charge-transfer excitations and reduces delocalization error in extended systems. |
| DFT+U / DFT+U+V | Adds Hubbard model corrections to specific orbitals (e.g., transition metal d, O p). Empirically penalizes delocalization, correcting electronic structure in strongly correlated materials. |
| Meta-GGA (e.g., SCAN) | Incorporates the kinetic energy density. Provides a more nuanced description of electron localization without empirical parameters, often reducing SIE. |
| Projector Augmented-Wave (PAW) Pseudopotentials | Provides access to full all-electron charge density and orbitals near the nucleus, crucial for accurate analysis of localized states and spin densities. |
| Density-Corrected DFT (DC-DFT) | Uses a more accurate density from a higher-level method in the XC functional evaluation. Directly targets delocalization error by correcting the input density. |
The Hohenberg-Kohn (HK) theorems establish that the ground-state electron density uniquely determines all properties of a many-electron system. This forms the theoretical bedrock of Density Functional Theory (DFT). However, the exact form of the exchange-correlation (XC) energy functional, E_XC[ρ], remains unknown. The choice of an approximate functional is therefore the most critical, and often most subjective, decision in a DFT simulation. This choice is heavily dependent on the system type—biological (soft, often involving weak interactions and large molecules) versus materials (hard, periodic, with strong bonding). This guide provides a structured decision framework within the broader context of HK theorem-based research, leveraging current benchmark data.
Core Functional Classes & Quantitative Performance
The following table summarizes key functional classes, their theoretical foundation, and quantitative performance metrics across benchmark sets for biological and materials systems. Data is compiled from recent sources including the GMTKN55 database for general main-group thermochemistry, kinetics, and non-covalent interactions, and materials-focused benchmarks.
Table 1: Functional Performance Across System Types
| Functional Class/Name | Key Ingredients (Exact Exchange %, DFT Correlation) | Typical Use Case | Biological System Performance* (Avg. Error) | Materials System Performance* (Avg. Error) | Notable Strengths | Notable Weaknesses |
|---|---|---|---|---|---|---|
| GGA (PBE) | GGA exchange, GGA correlation (0% HF) | Bulk materials, metals, periodic structures | Poor (Non-covalent int.: >2 kcal/mol) | Good (Lattice const.: ~1-2%) | Computationally cheap; good for metals, structural props. | Underbinds; poor for dispersion, reaction barriers. |
| Meta-GGA (SCAN) | Kinetic energy density (0% HF) | Diverse materials, surfaces | Moderate (Non-covalent int.: ~0.5-1 kcal/mol) | Very Good (Lattice const.: ~1%) | Good for diverse bonding without HF exchange. | Can be numerically sensitive; slower than GGA. |
| Hybrid GGA (PBE0, B3LYP) | Mixes GGA + HF exchange (20-25% HF) | Molecular electronics, band gaps, clusters | Good (Reaction barriers: ~2-3 kcal/mol) | Moderate (Band gaps: improves but underestimates) | Improved thermochemistry, band gaps over pure GGA. | Expensive for periodic systems; poor for pure metals. |
| Hybrid Meta-GGA (ωB97X-V, M06-2X) | Mixes Meta-GGA + HF exchange (high % HF, ~54-100%) | Biological molecules, drug design, spectroscopy | Excellent (Non-covalent int.: <0.5 kcal/mol) | Poor (Metallic systems) | Excellent for non-covalent interactions, thermochemistry. | Very expensive; parameterized; poor for extended systems. |
| Range-Separated Hybrid (HSE06) | Short-range: PBE0, Long-range: PBE (∼25% HF sr) | Semiconductors, band structure, defects | Moderate | Excellent (Band gaps: ~0.2-0.3 eV error) | Accurate band gaps; efficient for periodic systems. | Not for van der Waals bonds; parameterized screening. |
| Dispersion-Corrected (PBE-D3(BJ)) | PBE + Empirical dispersion correction | Molecular crystals, adsorption, organic materials | Very Good (Non-covalent int.: ~0.2-0.5 kcal/mol) | Very Good (Adsorption energies) | Adds crucial weak interactions cheaply. | Empirical; not a fundamental improvement to XC. |
Performance metrics are approximate average errors on relevant benchmark sets (e.g., S66 for non-covalent, CEDPG for solids). Errors are system-dependent. *HF = Hartree-Fock (Exact Exchange).
Decision Flowchart & Experimental Protocol Integration
The following diagram outlines the logical decision process for functional selection, integrated with the validation protocols required for each branch.
Title: Functional Selection Decision Flow
Validation Protocols:
- Protocol A (Materials, Structure/Energy): 1) Perform geometry optimization with PBE-D3. 2) Calculate cohesive energy (solids) or adsorption energy (surfaces). 3) Compare calculated lattice constants/equilibrium distances to experimental XRD data (tolerance: ±2%). 4) Validate phonon dispersion for dynamical stability.
- Protocol B (Materials, Meta-GGA Validation): 1) Optimize with SCAN. 2) Compute formation enthalpies for a test set of binary compounds. 3) Benchmark against experimental formation enthalpies (aim for MAE < 0.1 eV/atom). Use a finer k-point grid (≥ 12 × 12 × 12 for simple solids).
- Protocol C (Materials, Electronic Structure): 1) Perform PBE ground-state calculation. 2) Perform single-point HSE06 calculation on PBE geometry. 3) Compare indirect/direct band gaps to experimental optical absorption onset. 4) Analyze projected density of states (PDOS) for orbital character.
- Protocol D (Biological, High Accuracy): 1) Optimize molecular geometry with ωB97X-V/def2-TZVP. 2) Perform vibrational frequency analysis to confirm minima (no imaginary frequencies). 3) Calculate interaction energy for a known host-guest complex (e.g., from S66 benchmark). 4) Apply counterpoise correction for basis set superposition error (BSSE).
- Protocol E (Biological, Balanced): 1) Optimize with PBE0-D3/6-31G. 2) Perform higher-level single-point energy calculation with a larger basis set (e.g., def2-QZVP). 3) Calculate reaction energy profile for an enzymatic step (e.g., proton transfer).
- Protocol F (Biological, Legacy): 1) Use B3LYP-D3/6-31G* for direct comparison to prior literature. 2) Calculate NMR chemical shifts (via GIAO method) and compare to experimental spectra. 3) Perform time-dependent DFT (TD-DFT) calculation for UV-Vis spectra simulation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational "Reagents" for DFT Studies
| Item/Software (Solution) | Primary Function | Application Context |
|---|---|---|
| Gaussian, ORCA, Q-Chem | Quantum chemistry packages for molecular (biological) DFT. | High-accuracy hybrid DFT on finite systems; spectroscopy (NMR, IR), reaction pathways. |
| VASP, Quantum ESPRESSO, ABINIT | Plane-wave pseudopotential codes for periodic (materials) DFT. | Calculation of bulk/surface properties, electronic bands, density of states, defects. |
| def2-TZVP, 6-311++G, cc-pVTZ | High-quality Gaussian-type orbital (GTO) basis sets. | Accurate molecular calculations, especially for energies and non-covalent interactions. |
| PAW Pseudopotentials (VASP), ONCVPSP | Projector Augmented-Wave and norm-conserving pseudopotentials. | Efficient plane-wave calculations replacing core electrons, crucial for heavy elements. |
| D3, D3(BJ), vdW-DF | Dispersion correction schemes. | Adding long-range van der Waals interactions to GGAs/meta-GGAs for organic/biomolecules and molecular crystals. |
| MongoDB/FireWorks | Workflow management databases. | Automating and tracking high-throughput computational screening of materials/drug candidates. |
| Pymatgen, ASE | Python libraries for materials/atomic scale analysis. | Generating input structures, analyzing output files (energies, structures), and creating post-processing scripts. |
| CHELPG, Hirshfeld | Atomic charge partitioning schemes. | Deriving partial atomic charges for analyzing electrostatic potentials in drug binding sites or surfaces. |
Advanced Pathway: Integrating DFT with Multi-Scale Models
For biological systems, DFT often informs parameters for larger-scale models. The following workflow depicts this integration for a drug-receptor binding study.
Title: QM/MM Multi-Scale Drug Binding Workflow
Selecting the correct functional is not a one-size-fits-all process but a hypothesis-driven decision that must align with the system's physics and the property of interest. For materials systems, SCAN and HSE06 represent the modern meta-GGA and hybrid standards for structural and electronic properties, respectively, often augmented with D3 corrections. For biological systems, range-separated hybrid meta-GGAs (ωB97X-V) offer state-of-the-art accuracy for interactions and spectroscopy. All choices must be validated against appropriate experimental or high-level theoretical benchmarks, as outlined in the provided protocols. This structured approach, grounded in the foundational Hohenberg-Kohn theorems, ensures that the approximation to E_XC[ρ] serves as a tool for discovery rather than a source of error.
The Hohenberg-Kohn (HK) theorems establish the existence of a unique one-to-one mapping between the ground-state electron density of a many-body system and its external potential. This foundational principle of Density Functional Theory (DFT) reduces the 3N-dimensional many-electron wavefunction problem to a 3-dimensional electron density problem. However, the practical application of DFT requires two critical approximations: the exchange-correlation functional and the representation of the Kohn-Sham orbitals. This guide focuses on the latter, specifically the dual challenges of basis set convergence—how to represent these orbitals with a finite set of functions—and the use of pseudopotentials (or effective core potentials, ECPs) to replace core electrons, thereby reducing computational cost. The central thesis is that optimal computational materials science and drug discovery require a meticulous, system-aware balance between these two factors to achieve chemically accurate results at feasible computational expense.
Theoretical Foundations and Definitions
Basis Set: A set of mathematical functions (e.g., Gaussian-type orbitals, plane waves) used to expand the Kohn-Sham orbitals. Completeness is approached as the basis set size increases. Pseudopotential: A simplified potential that replaces the strong Coulomb potential and inert core electrons of an atom, modeling only the chemically active valence electrons. It incorporates relativistic effects and projects out core states.
Basis Set Convergence: Methodology and Quantitative Benchmarks
Achieving convergence in calculated properties with respect to basis set size is non-trivial. The required level depends on the target property.
Experimental Protocol for Convergence Testing
- System Selection: Choose a representative molecular or solid-state system relevant to the research (e.g., a drug fragment, a catalyst cluster).
- Property Definition: Define the target property: Total Energy, Atomization Energy, Reaction Barrier Height, Bond Length, Vibrational Frequency, or Interaction Energy (e.g., binding affinity).
- Basis Set Hierarchy: Perform a series of single-point energy (or geometry optimization) calculations using a systematically improvable sequence of basis sets (e.g., for Gaussian-type orbitals: Pople series
6-31G*→6-311+G→aug-cc-pVDZ→aug-cc-pVTZ→aug-cc-pVQZ; for plane waves: increasing cutoff energy). - Reference Establishment: Use the largest, computationally feasible basis set or a complete basis set (CBS) extrapolation result as a reference.
- Error Analysis: Calculate the absolute or relative error of the target property for each basis set relative to the reference. Plot error vs. basis set size/cost.
Quantitative Data on Convergence Rates
Table 1: Typical Convergence of Properties for Organic Molecules with Gaussian-Type Basis Sets
| Property | Convergence Speed | Min. Recommended Basis | Error at aug-cc-pVDZ |
Error at aug-cc-pVTZ |
|---|---|---|---|---|
| Total Energy | Very Slow | aug-cc-pVQZ or larger |
~1000 kJ/mol | ~100 kJ/mol |
| Atomization Energy | Moderate | aug-cc-pVTZ |
~10-20 kJ/mol | ~1-4 kJ/mol |
| Molecular Geometry | Fast | cc-pVDZ |
~0.01 Å (bond length) | ~0.001 Å |
| Reaction Energy | Moderate | aug-cc-pVTZ |
~8-15 kJ/mol | ~2-4 kJ/mol |
| Vibrational Frequencies | Fast-Moderate | cc-pVTZ |
~20-50 cm⁻¹ | ~5-10 cm⁻¹ |
Table 2: Plane-Wave Cutoff Energy Convergence for a Semiconductor (e.g., Silicon)
| Property | Cutoff (eV) | Total Energy Error (meV/atom) | Band Gap Error (eV) | Lattice Constant Error (Å) |
|---|---|---|---|---|
| Reference | 800 | 0.0 | 0.00 | 0.000 |
| Fast / Low Accuracy | 300 | 25.4 | 0.45 | 0.025 |
| Balanced | 500 | 5.1 | 0.12 | 0.008 |
| High Accuracy | 700 | 0.8 | 0.03 | 0.002 |
Pseudopotentials: Types, Generation, and Validation Protocols
Types and Applications
- Norm-Conserving (NC): Strictly preserve charge density outside core radius. Require moderate plane-wave cutoffs. Good for general-purpose solid-state calculations.
- Ultrasoft (US): Allow a softer, more computationally efficient potential by relaxing the norm-conserving condition. Require lower cutoffs.
- Projector Augmented-Wave (PAW): A rigorous, all-electron reconstruction method that uses auxiliary projector functions. Considered the state-of-the-art for accuracy vs. cost in plane-wave codes.
Protocol for Pseudopotential Validation
- Generation/Selection: Choose a library (e.g.,
SSSP,GBRV,SG15) or generate a pseudopotential with defined core-valence partitioning, exchange-correlation functional, and generation code. - All-Electron Reference: Perform an all-electron calculation (e.g., using the FHI-AIMS code with tight numerical settings) for a set of test structures (atom, dimer, bulk).
- Test Suite Calculation: Use the pseudopotential to compute the same test suite.
- Property Comparison: Benchmark key properties against the all-electron result.
- Primary Properties: Equation of state (lattice constant, bulk modulus), cohesive energy, electronic band structure.
- Secondary Properties: Phonon frequencies, elastic constants.
- Tolerance Check: Ensure errors fall within acceptable chemical accuracy limits (e.g., < 1 meV/atom for energy, < 0.01 Å for structure).
Quantitative Data on Pseudopotential Accuracy
Table 3: Benchmark of Pseudopotential Types for Gold (Au) Bulk Phase
| PP Type | XC Functional | Cutoff (eV) | Δ Lattice Const. (Å) | Δ Cohesive Energy (meV/atom) | Δ Bulk Modulus (GPa) | Relative Speed |
|---|---|---|---|---|---|---|
| All-Electron | PBE | N/A | 0.000 (Ref) | 0 (Ref) | 0 (Ref) | 1.0x (Baseline) |
| NC (FR) | PBE | 800 | +0.010 | +15 | -1.5 | 3.2x |
| US (GBRV) | PBE | 500 | -0.005 | -8 | +0.8 | 5.1x |
| PAW (VASP) | PBE | 700 | +0.002 | +2 | -0.2 | 2.5x |
The Integrated Workflow: Balancing Factors
The optimal choice is system- and property-dependent. A drug developer modeling protein-ligand binding requires excellent treatment of dispersion (often needing large basis sets) and transition metals (requiring high-quality pseudopotentials).
Decision Pathway for Method Selection
Diagram 1: Decision Tree for Basis Set and PP Selection (96 chars)
Convergence-Pseudopotential Interplay Diagram
Diagram 2: Cost vs. Accuracy Interplay in DFT Setup (89 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Computational "Reagents" for DFT Studies
| Item / Solution | Function / Purpose | Example Sources / Libraries |
|---|---|---|
| Gaussian-Type Basis Sets | Expand molecular orbitals in quantum chemistry codes. Provide systematic improvability. | Pople series, Dunning (cc-pVXZ), Karlsruhe (def2-XVP) |
| Plane-Wave Pseudopotential Libraries | Provide pre-validated, consistent pseudopotentials for plane-wave DFT calculations. | SSSP (Standard Solid State Pseudopotentials), SG15, GBRV, PseudoDojo |
| All-Electron Reference Data | Serve as the "ground truth" for validating pseudopotentials and basis set convergence. | NIST CCCBDB, Materials Project, FHI-AIMS results |
| CBS Extrapolation Formulas | Mathematical formulas to estimate the Complete Basis Set limit from calculations with two or more basis set sizes. | 1/X³ (for HF energy), exponential (for correlation energy) |
| Basis Set Superposition Error (BSSE) Correction | Corrects for artificial stabilization in interaction energies due to fragment basis set incompleteness. | Counterpoise (Boys-Bernardi) correction scheme |
| Pseudopotential Generation Codes | Software to create consistent, tunable pseudopotentials for specific elements and configurations. | ONCVPSP, APE, ATOM (included with Quantum ESPRESSO) |
The pursuit of accurate and computationally efficient DFT simulations necessitates a deep understanding of the trade-offs between basis set completeness and pseudopotential approximation. Grounded in the Hohenberg-Kohn framework, the choice is not about seeking a single "best" option but about making an informed, property-specific compromise. For high-accuracy studies of molecular systems (e.g., drug design), all-electron calculations with correlation-consistent basis sets (aug-cc-pVTZ or higher) are often required. For materials science and high-throughput screening, modern PAW or ultrasoft pseudopotentials from validated libraries, combined with a carefully tested plane-wave cutoff, provide the best balance. The mandatory step for any serious research is a preliminary convergence study—varying both the basis set/cutoff and testing different pseudopotentials for the key elements involved—to establish a protocol that delivers the required accuracy at minimal computational expense.
The Hohenberg-Kohn (HK) theorems establish the foundational principle that the ground-state electron density uniquely determines all properties of a many-electron system. However, the exact universal functional, F[ρ], remains unknown. In biomolecular systems, a critical component of this functional is the description of non-covalent, weak interactions—primarily van der Waals (vdW) or dispersion forces. These forces are quantum mechanical in origin, arising from correlated fluctuations of electron densities, and are not captured by standard local or semi-local density functional approximations (LDA, GGA). Their accurate inclusion is paramount for predicting protein-ligand binding affinities, protein folding, and macromolecular assembly, directly impacting structure-based drug design. This guide details modern corrective schemes for incorporating vdW interactions within the Kohn-Sham density functional theory (KS-DFT) framework, contextualizing them as essential, a posteriori approximations to the exact HK functional for systems where electron correlation is long-ranged.
Core vdW-Corrective Schemes: Mechanisms and Implementation
The following table categorizes and summarizes the principal corrective approaches.
Table 1: Summary of Prominent vdW-Corrective Schemes for Biomolecular Systems
| Scheme Category | Specific Method | Key Functional Form / Parameterization | System Type (Typical Use) | Computational Scaling | Key Strengths | Key Limitations |
|---|---|---|---|---|---|---|
| Pairwise Energy Correction | DFT-D3 (Grimme et al.) | $E{disp} = -\sum{A>B}\sum{n=6,8}sn \frac{C^{AB}n}{r{AB}^n} f{damp}(r{AB})$ | Large biomolecules, supramolecular systems | O(N²) | Highly efficient, system-specific damping, geometry-dependent coefficients. | Non-local correlation effects not captured. |
| Pairwise Energy Correction | DFT-D4 | Similar to D3, with atomic partial charge-dependent coefficients. | As above, improved for diverse elements. | O(N²) | Improved charge sensitivity and reference data. | Same fundamental pairwise limitation. |
| Non-Local Correlation Functional | vdW-DF (Dion et al.) | $Ec^{nl} = \frac{1}{2}\iint d\mathbf{r} d\mathbf{r}' n(\mathbf{r}) \phi(q, q', r{12}) n(\mathbf{r}')$ | Surfaces, layered materials, binding in aqueous pockets. | O(N² log N) | Seamless integration, no empirical atom typing. | Can overestimate binding distances; early versions underbind. |
| Non-Local Correlation Functional | VV10 (Vydrov & Van Voorhis) | $Ec^{nl} = \int d\mathbf{r} n(\mathbf{r}) \epsilonc^{nl}(\mathbf{r})$ with a double integral kernel. | Broad, including soft and hard matter. | O(N² log N) | Good accuracy across many system types. | Parameter $\omega$ needs optimization for specific functionals. |
| Exchange-Hole Dipole Moment | XDM (Becke & Johnson) | Models dispersion from the exchange-hole dipole moment. | Small molecule crystals, medium-sized clusters. | O(N³) (depends on functional) | Derivable from the electron density. | Less tested on very large, flexible biomolecules. |
Experimental & Computational Validation Protocols
The efficacy of vdW corrections is benchmarked against high-level quantum chemical calculations and experimental data.
Protocol for Benchmarking Binding Affinities (S66x8 Database)
- System Preparation: Extract the 66 biologically relevant molecular dimer structures (e.g., hydrogen-bonded, dispersion-dominated, mixed) from the S66x8 dataset. Generate the 8 geometries per dimer (original equilibrium and seven displaced).
- Electronic Structure Calculations:
- Reference: Perform highly accurate CCSD(T) calculations at the complete basis set (CBS) limit using, for example, the ORCA or MRCC software. This serves as the benchmark.
- Test: Perform KS-DFT calculations with and without the vdW corrective scheme under test. Use a consistent medium-sized basis set (e.g., def2-TZVP). Employ software like Quantum ESPRESSO, VASP, or Gaussian.
- Data Analysis: For each dimer geometry, compute the interaction energy ($E{int} = E{dimer} - E{monomerA} - E{monomerB}$). Calculate the mean absolute error (MAE) and root mean square error (RMSE) relative to the CCSD(T)/CBS benchmark across all 528 data points. Tabulate results separately for different interaction types.
Protocol for Protein-Ligand Binding Free Energy (ΔG) Calculation
- System Setup: Obtain a high-resolution crystal structure of a protein-ligand complex (e.g., from the PDB). Prepare the system using standard molecular dynamics protocols (protonation, solvation in explicit water, ion neutralization) with tools like
tleap(AmberTools) orCHARMM-GUI. - Hybrid QM/MM Simulations: Employ a QM/MM partitioning scheme. The ligand and key protein residues (e.g., active site) are treated with vdW-corrected DFT (QM region). The remainder is treated with a classical force field (MM region).
- Free Energy Perturbation (FEP) or Thermodynamic Integration (TI): Use alchemical transformation methods to compute the absolute or relative binding free energy. Perform extensive sampling (multiple nanoseconds) using MD engines like OpenMM or NAMD interfaced with a QM code (e.g., CP2K). The vdW correction is active in the QM region throughout.
- Validation: Compare the calculated ΔG with experimentally measured inhibition constants (Ki) or dissociation constants (Kd) from isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR).
Visualization of Conceptual and Workflow Relationships
Title: The Role of vdW Corrections in KS-DFT from HK Theorems
Title: Workflow for Validating vdW-Corrective Schemes
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Tools and Resources for vdW-Corrected Biomolecular Simulations
| Item / Resource | Function / Role | Example (Vendor / Source) |
|---|---|---|
| Quantum Chemistry Software | Performs the core electronic structure calculation with implemented vdW corrections. | CP2K (Open Source), Quantum ESPRESSO (OS), VASP (Commercial), Gaussian (Com.), ORCA (Free for Acad.) |
| Classical MD Engine | Handles system preparation, force field-based dynamics, and hybrid QM/MM integration. | GROMACS (OS), NAMD (OS), AMBER (Com.), CHARMM (Com.), OpenMM (OS) |
| Benchmark Databases | Provides curated sets of high-quality reference data for validation. | S66x8, L7, HALogen (from Hobza group); BioFragment (Sherrill group) |
| Force Fields with vdW | Provides classical parameters for dispersion, often refined against QM data. | CHARMM36 (with LJ terms), AMBER ff19SB, OPLS4, GAFF2 |
| Analysis & Visualization | Analyzes trajectories, calculates energies, and visualizes molecular structures. | VMD, PyMOL, MDAnalysis (Python), CPPTRAJ |
| High-Performance Compute (HPC) Cluster | Provides the necessary computational power for QM and QM/MM calculations. | Local university clusters, XSEDE resources, AWS/GCP/Azure cloud HPC instances |
The Hohenberg-Kohn (HK) theorems establish the theoretical foundation for Density Functional Theory (DFT), proving that the ground-state electron density uniquely determines all properties of a many-electron system. While this provides immense conceptual simplification, practical computational implementations introduce significant challenges. The central thesis of this guide is that rigorous management of convergence issues and computational parameters is not merely a technical detail but a fundamental requirement for obtaining physically meaningful, reliable, and reproducible results from DFT calculations—results that genuinely reflect the predictions of the HK theorems. Inaccurate convergence can lead to erroneous electron densities, violating the core tenet of the theory.
Core Convergence Parameters in DFT Calculations
The following table summarizes the primary computational parameters requiring systematic convergence testing to ensure the calculated electron density and derived properties are independent of numerical discretization.
Table 1: Key Computational Parameters and Convergence Criteria
| Parameter | Description | Typical Convergence Target | Impact on Results |
|---|---|---|---|
| Energy Cutoff (E_cut / ENMAX) | Plane-wave kinetic energy cutoff for basis set. | ΔE < 1 meV/atom | Total energy, forces, stress, electronic structure. |
| k-point Mesh Density | Sampling of the Brillouin Zone. | ΔE < 1 meV/atom | Band structure, density of states, total energy. |
| Electronic Step Convergence (EDIFF) | Tolerance for SCF cycle. | EDIFF < 1E-5 eV | Accuracy of self-consistent electron density. |
| Force Convergence (EDIFFG) | Tolerance for ionic relaxation. | EDIFFG < 0.01 eV/Å | Equilibrium geometry, phonon frequencies. |
| Density Mixing Parameters | Algorithm for SCF charge density mixing. | Stable SCF in < 50 cycles | SCF convergence stability, avoidance of charge sloshing. |
| Smearing Width (SIGMA) | Electronic occupancy smearing for metals. | Entropy term (TS) < 0.1 meV/atom | Accurate metallic states, total energy of conductors. |
Experimental Protocol: A Systematic Convergence Study
This protocol outlines a robust methodology for establishing converged parameters for a DFT study of a new material or molecule, ensuring reliability and reproducibility.
Protocol Title: Systematic Convergence Testing for DFT-Based Property Prediction
Objective: To determine a set of computationally efficient yet sufficiently accurate parameters for which key physical properties (total energy, lattice constants, band gap) are invariant to further increases in numerical precision.
Materials & Computational Setup:
- Software: VASP, Quantum ESPRESSO, or equivalent ab initio package.
- Functional: Select exchange-correlation functional (e.g., PBE, HSE06).
- Pseudopotential: Choose appropriate projector-augmented wave (PAW) or norm-conserving potentials.
- Initial Structure: Provide crystallographic information file (CIF) or molecular coordinate file.
Procedure:
Baseline Establishment:
- Start with standard recommended parameters for the chosen pseudopotential.
- Perform a coarse structural relaxation to obtain a stable starting geometry.
Energy Cutoff Convergence:
- Fix a moderate k-point mesh.
- Perform single-point energy calculations for the relaxed structure, incrementally increasing the plane-wave energy cutoff (e.g., 300, 400, 500, 600 eV).
- Plot total energy per atom versus cutoff energy. The converged value is where the energy change is less than the target (1 meV/atom).
k-point Mesh Convergence:
- Fix the energy cutoff at the converged value from Step 2.
- Perform single-point calculations with increasingly dense k-point meshes (e.g., 2x2x2, 3x3x3, 4x4x4, 5x5x5 for a cubic system).
- Plot total energy per atom versus k-point density. Identify the mesh where energy change is below the target.
Simultaneous Verification:
- Perform a final check using the determined E_cut and k-mesh on a secondary property of interest (e.g., lattice constant, band gap). Confirm this property is also stable.
SCF and Relaxation Convergence:
- Using the converged basis set, systematically tighten the EDIFF and EDIFFG tolerances to ensure forces and geometry are fully relaxed. Monitor the number of electronic and ionic steps required.
Data Analysis:
- Generate convergence plots for each parameter.
- The final parameter set is defined as the point of diminishing returns, where computational cost increases significantly without meaningful change in output properties.
- This complete parameter set must be explicitly documented in any publication to ensure reproducibility.
Workflow and Logical Relationships
Diagram Title: The Critical Path from Theory to Reproducible DFT Results
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Computational "Reagents" and Materials for DFT Studies
| Item / "Reagent" | Function & Purpose | Example / Note |
|---|---|---|
| Exchange-Correlation Functional | Approximates quantum mechanical electron-electron interactions; the primary "ingredient" defining the physical model. | PBE (general purpose), HSE06 (hybrid, for band gaps), SCAN (meta-GGA, improved accuracy). |
| Pseudopotential / PAW Dataset | Replaces core electrons with an effective potential, reducing computational cost while retaining valence electron accuracy. | VASP PAW libraries, SSSP (Standard Solid State Pseudopotentials) efficiency/accuracy sets. |
| Plane-Wave Basis Set | The mathematical set of functions used to expand the electron wavefunctions and density. | Defined by the kinetic energy cutoff (ENMAX). Quality is tied to the pseudopotential. |
| k-point Sampling Scheme | Discretizes continuous Brillouin Zone for numerical integration over electronic states. | Monkhorst-Pack grids, Gamma-centered meshes. Density crucial for metals and semiconductors. |
| SCF Convergence Algorithm | Solves the non-linear Kohn-Sham equations iteratively to find a self-consistent electron density. | RMM-DIIS, Pulay mixing, Kerker preconditioner (for metallic systems). |
| Ionic Relaxation Algorithm | Finds the minimum-energy atomic configuration (ground-state geometry). | Conjugate gradient, quasi-Newton (BFGS), damped molecular dynamics (FIRE). |
| High-Performance Computing (HPC) Cluster | Provides the necessary computational power to perform calculations with converged parameters in a reasonable time. | CPU/GPU nodes, fast interconnects, sufficient memory per core. |
Benchmarking DFT: How It Stacks Up Against Wavefunction and Machine Learning Methods
The Hohenberg-Kohn (HK) theorems establish the theoretical foundation for Density Functional Theory (DFT), proving that the ground-state electron density uniquely determines all properties of a many-electron system. This elegant formalism reduces the 3N-dimensional many-body wavefunction problem to a 3-dimensional electron density problem, promising a computationally tractable path to accurate quantum mechanical calculations. The first HK theorem validates the existence of a density functional for the energy, while the second provides a variational principle. In practice, the exact functional is unknown, leading to the development of approximate exchange-correlation (XC) functionals (LDA, GGA, meta-GGAs, hybrids). This stands in contrast to Post-Hartree-Fock (Post-HF) methods, which systematically improve upon the mean-field Hartree-Fock solution by adding explicit treatments of electron correlation via wavefunction-based approaches, such as Møller-Plesset perturbation theory (MP2) or coupled-cluster theory (CCSD(T)). The central trade-off in computational chemistry is between the formally exact but unknown XC functional in DFT and the systematically improvable but combinatorially scaling Post-HF methods.
Fundamental Methodologies and Computational Scaling
Density Functional Theory (DFT): DFT solves the Kohn-Sham equations, which map the interacting system of electrons onto a fictitious system of non-interacting electrons moving in an effective potential. The computational cost is dominated by handling the Kohn-Sham orbitals, leading to formal scaling of O(N³) for the diagonalization step, where N is proportional to the number of basis functions. With efficient implementations and linear-scaling techniques for large systems, effective scaling can approach O(N) for certain problems.
Post-Hartree-Fock Methods:
- MP2 (Second-Order Møller-Plesset Perturbation Theory): Adds electron correlation via Rayleigh-Schrödinger perturbation theory. It is non-iterative and scales formally as O(N⁵), where N is the number of basis functions.
- CCSD(T) (Coupled-Cluster Singles, Doubles, and perturbative Triples): The "gold standard" for molecular energetics. The coupled-cluster equations are solved iteratively. CCSD scales as O(N⁶), and the perturbative (T) correction adds O(N⁷) scaling.
Quantitative Comparison of Accuracy and Cost
Table 1: Formal Scaling and Typical Application Range
| Method | Formal Computational Scaling | Typical System Size (Atoms) | Typical Accuracy (kJ/mol) |
|---|---|---|---|
| DFT (GGA/Hybrid) | O(N³) | 10s - 1000s | 10 - 40 |
| MP2 | O(N⁵) | 10s - 100s | 5 - 20 |
| CCSD(T) | O(N⁷) | < 50 | < 4 |
Table 2: Performance on Benchmark Sets (e.g., GMTKN55, S66)
| Property | Best DFT Functionals (e.g., ωB97M-V) | MP2 (with correction) | CCSD(T) [Reference] |
|---|---|---|---|
| Non-covalent Interactions | ~2-5% MAE | ~2-4% MAE (with D3) | 0% MAE (by definition) |
| Reaction Barriers | ~4-8% MAE | ~3-6% MAE | ~1% MAE |
| Transition Metal Energetics | Highly variable (10-40% MAE) | Often fails | Required for benchmark |
Experimental Protocol for Benchmarking:
- System Selection: Choose a diverse benchmark set (e.g., S66 for non-covalent interactions, Diels-Alder reactions for barriers).
- Geometry Optimization: Optimize all structures using a high-level method (e.g., DFT with a tight functional) and a large basis set.
- Single-Point Energy Calculation: Perform high-level energy evaluations on the fixed geometries.
- DFT Protocol: Use a range of XC functionals (PBE, B3LYP, ωB97X-D, etc.) with a large triple-zeta basis set (e.g., def2-TZVP) and appropriate dispersion correction (D3(BJ)).
- MP2 Protocol: Use the same basis set. Apply an additive correction for basis set superposition error (BSSE) via the counterpoise method. Consider spin-component scaling (SCS-MP2) or explicitly correlated MP2-F12 to accelerate basis set convergence.
- CCSD(T) Protocol: Perform CCSD(T) calculations with a correlation-consistent basis set (e.g., cc-pVTZ). Apply extrapolation to the complete basis set (CBS) limit. Use this result as the reference "truth" for calculating errors of other methods.
- Error Analysis: Calculate Mean Absolute Errors (MAE), Root-Mean-Square Errors (RMSE), and maximum deviations relative to the CCSD(T)/CBS reference.
Diagram: Method Selection Workflow
Diagram Title: Quantum Chemistry Method Decision Tree
The Scientist's Toolkit: Essential Computational Reagents
Table 3: Key Research Reagent Solutions
| Item/Software | Category | Function/Brief Explanation |
|---|---|---|
| Gaussian, ORCA, Q-Chem, PySCF | Quantum Chemistry Packages | Integrated suites to perform DFT, MP2, CCSD(T), and other calculations with various basis sets and functionals. |
| def2-TZVP, cc-pVTZ, 6-311+G(d,p) | Gaussian-Type Basis Sets | Sets of mathematical functions (orbitals) used to expand molecular orbitals. Choice balances accuracy and cost. |
| D3(BJ) Correction | Empirical Dispersion Correction | Adds van der Waals dispersion forces, crucial for DFT accuracy in non-covalent interactions. |
| RI/DF Approximation | Density Fitting | Approximates electron repulsion integrals, drastically speeding up MP2 and hybrid DFT calculations. |
| DLPNO Approximation | Local Correlation Method | ("Domain-based Local Pair Natural Orbital") Enables CCSD(T)-level calculations on systems with hundreds of atoms by truncating correlations to local domains. |
| CP2K, FHI-aims | Periodic DFT Codes | Specialized for extended solid-state systems and large-scale molecular dynamics simulations. |
| GMTKN55 Database | Benchmark Suite | A collection of 55 datasets for evaluating the general accuracy of quantum chemical methods across diverse chemical problems. |
Diagram: Logical Relationship of Ab Initio Methods
Diagram Title: Hierarchy of Electronic Structure Methods
The choice between DFT and Post-HF methods is dictated by the specific interplay between desired accuracy, system size, and available computational resources. For drug development professionals studying large biomolecules, DFT with robust dispersion corrections is the indispensable workhorse. For researchers developing highly accurate force fields or validating mechanisms for small catalytic systems, CCSD(T) remains the indispensable benchmark. Modern developments are blurring these trade-offs: local correlation approximations (e.g., DLPNO-CCSD(T)) are bringing "gold standard" accuracy to larger systems, while machine-learned density functionals promise to embed high-level correlation at DFT cost. Ultimately, these methods are complementary, with Post-HF providing the benchmarks necessary to guide the development of more reliable and broadly applicable density functionals, fulfilling the promise of the Hohenberg-Kohn theorems.
Within the rigorous framework established by the Hohenberg-Kohn (HK) theorems, which confirm the existence of a unique energy functional of the electron density, density functional theory (DFT) provides the foundation for modern electronic structure calculations. The central challenge is the unknown exact exchange-correlation (XC) functional. "Jacob's Ladder" provides a systematic, hierarchical metaphor for classifying XC functionals, where each rung represents an increase in complexity and the incorporation of more physical ingredients, with the goal of reaching "chemical accuracy" (~1 kcal/mol error). This whitepaper details the rungs of the ladder, provides quantitative comparisons, outlines experimental validation protocols, and presents essential research tools.
The two Hohenberg-Kohn theorems form the non-empirical bedrock of DFT. The first theorem proves that the ground-state electron density uniquely determines the external potential (and thus all properties of the system). The second theorem provides a variational principle: the exact ground-state density minimizes the total energy functional. This establishes DFT as a formally exact theory. However, the universal energy functional E[ρ] is divided into known parts (kinetic energy of non-interacting electrons, electron-nuclear attraction, classical Coulomb repulsion) and one critical unknown: the exchange-correlation functional EXC[ρ]. The pursuit of accurate approximations to EXC[ρ] is the ascent of Jacob's Ladder.
The Rungs of Jacob's Ladder
The ladder is conceptualized as follows, with each higher rung incorporating more "ingredients" from the wavefunction, moving beyond the pure density.
Diagram Title: Hierarchy of Jacob's Ladder in DFT
Quantitative Comparison of Functional Rungs
The following table summarizes key functionals, their ingredients, computational cost, and typical accuracy for thermochemical properties.
Table 1: Characteristics of Representative Functionals on Jacob's Ladder
| Rung | Functional Class | Key Ingredient(s) | Example Functionals | Mean Absolute Error (MAE) on G3/99 Set (kcal/mol) | Typical Computational Cost Scaling |
|---|---|---|---|---|---|
| 1 | Hartree-Fock | Exact Exchange | HF | ~120-150 | O(N⁴) |
| 2 | LSDA | Local Density ρ | SVWN | ~35-40 | O(N³) |
| 3 | GGA | ρ, ∇ρ | PBE, BLYP | ~7-10 | O(N³) - O(N⁴) |
| 4 | Meta-GGA | ρ, ∇ρ, τ (kinetic energy density) | SCAN, TPSS | ~4-6 | O(N⁴) |
| 5 | Hybrid | ρ, ∇ρ, Exact Exchange Mix | B3LYP, PBE0 | ~3-5 | O(N⁴) - O(N⁵) |
| 5+ | Double Hybrid | ρ, ∇ρ, Exact Exchange + MP2 Correlation | B2PLYP, DSD-PBEP86 | ~1-2 | O(N⁵) |
Data sourced from recent benchmark studies (2022-2024). The G3/99 set is a standard benchmark of thermochemical data. Cost scaling is with system size N.
Experimental Validation Protocols
Achieving chemical accuracy requires rigorous benchmarking against high-quality experimental and theoretical reference data.
Protocol for Benchmarking Thermochemical Accuracy
Objective: To determine the mean absolute error (MAE) and maximum deviation of a functional for atomization energies, ionization potentials, electron affinities, and proton affinities.
Materials: Standard benchmark sets (e.g., GMTKN55, MG8, ROST61). Software: Quantum chemistry packages (e.g., ORCA, Gaussian, Q-Chem, PySCF). Hardware: High-performance computing cluster.
Method:
- System Selection: Obtain molecular geometries for all species in the chosen benchmark set from databases like the NIST Computational Chemistry Comparison and Benchmark Database (CCCBDB).
- Calculation Setup:
- Perform single-point energy calculations on each species using the target functional and a consistent, high-quality basis set (e.g., def2-QZVP).
- Critical: Apply a consistent treatment of dispersion corrections (e.g., D3(BJ)) if the base functional lacks it.
- Use tight convergence criteria for the self-consistent field (SCF) and geometry.
- Energy Composition: For properties like atomization energy, calculate: Eatomization = Σ E(atoms) - E(molecule) using the same functional and basis set for all components.
- Error Analysis: Compare calculated values to the trusted reference values from the benchmark set. Compute MAE, root-mean-square error (RMSE), and maximum error.
- Statistical Reporting: Present errors disaggregated by chemical property subset (e.g., organic vs. inorganic, bonded vs. non-covalent).
Protocol for Validating Non-Covalent Interactions
Objective: To assess functional performance for weak interactions (hydrogen bonds, dispersion, π-π stacking).
Materials: S66, S30L, or NCI benchmark sets. Method:
- Use counterpoise-corrected interaction energy calculations to eliminate basis set superposition error (BSSE).
- For each dimer in the set, calculate: Eint = E(AB)dimer geometry - E(A)dimer geometry - E(B)dimer geometry where single-point energies for monomers A and B are computed using the dimer's basis set but with the ghost orbital method.
- Compare to highly accurate reference data (often from CCSD(T)/CBS calculations).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for DFT Research
| Item / Solution | Function / Purpose | Key Considerations for Selection |
|---|---|---|
| Software Packages | Provides the engine for SCF cycles, integral computation, and functional implementation. | ORCA (free, extensive), Gaussian (industry standard), Q-Chem (advanced functionals), VASP (solid-state). |
| Basis Sets | Mathematical functions representing atomic orbitals. | def2-series (balanced), cc-pVXZ (correlation consistent), plane-waves (periodic systems). Choice balances accuracy and cost. |
| Pseudopotentials/PPs | Replace core electrons for heavier atoms, reducing cost. | CRENBL, SBKJC. Must be compatible with chosen functional and basis set. |
| Dispersion Corrections | Add London dispersion forces missing in many lower-rung functionals. | Grimme's D3(BJ) with damping is standard. Essential for non-covalent interactions. |
| Benchmark Databases | Source of reliable reference data for validation. | NIST CCCBDB, GMTKN, MoITor. Critical for assessing functional performance. |
| High-Performance Computing (HPC) | Provides the computational power for large systems/high-level calculations. | Access to CPU/GPU clusters is necessary for production research on drug-sized molecules. |
Pathway to Chemical Accuracy: A Logical Workflow
The following diagram illustrates the decision pathway a researcher might follow when selecting a functional for a drug development application, balancing accuracy and computational cost.
Diagram Title: DFT Functional Selection Workflow for Drug Research
The ascent of Jacob's Ladder, grounded in the exact HK theorems, represents a concerted effort to incorporate increasingly sophisticated physical insights into the exchange-correlation functional. While higher rungs (meta-GGAs, hybrids, double hybrids) systematically approach chemical accuracy, they do so at increased computational cost. For researchers in drug development, the choice of functional is not merely a selection from a menu but a strategic decision based on the target property (e.g., binding affinity vs. reaction barrier), system size, and available resources. The ongoing development of functionals, particularly on the fourth and fifth rungs, continues to narrow the gap between efficient DFT calculations and benchmark quantum chemical accuracy, solidifying its role as an indispensable tool in computational chemistry and pharmaceutical design.
The foundational theorems of density functional theory (DFT), established by Hohenberg and Kohn, state that all ground-state electronic properties of a system are uniquely determined by its electron density. This forms the theoretical bedrock for modern computational predictions of molecular properties, including redox potentials (E⁰) and pKa values. In biomedicine, accurate prediction of these parameters is critical for understanding drug metabolism, antioxidant activity, enzyme mechanism, and the design of redox-active therapeutics. However, the translation of the exact, non-relativistic Hohenberg-Kohn theorems into practical, approximate functionals for complex, solvated biomolecules presents significant validation challenges. This whitepaper examines the state of the art, detailing methodologies, successes, persistent challenges, and experimental protocols for validation.
Quantitative Data on Prediction Accuracy
Table 1: Performance of Computational Methods for pKa and Redox Potential Prediction
| Property | System Type | Method/Functional | Basis Set/Solvent Model | Mean Absolute Error (MAE) | Key Challenge |
|---|---|---|---|---|---|
| pKa | Small organic molecules | Direct ΔG (DFT) | 6-31+G(d), SMD | ~1.5 - 3.0 pKa units | Solvation energy, proton solvation energy |
| pKa | Protein residues (e.g., Asp, Glu) | Empirical FEP/MC | Poisson-Boltzmann, MCCE | ~0.5 - 1.2 pKa units | Dielectric environment, conformational sampling |
| pKa | Drug-like molecules (multiprotic) | QM/MM, COSMO-RS | B3LYP/6-31G(d), COSMO | ~0.8 - 2.0 pKa units | Multiple microstates, tautomerism |
| Redox Potential (E⁰) | Quinones (in solution) | DFT (B3LYP, M06-2X) | 6-311++G(2d,2p), IEFPCM | ~50 - 150 mV | Solvent reorganization, functional dependence |
| Redox Potential (E⁰) | Heme proteins (Cytochromes) | DFT+Poisson-Boltzmann | OPBE, TZP, CPCM | ~30 - 80 mV | Spin state, axial ligand effects, protein polarization |
| Redox Potential (E⁰) | Flavoproteins | QM/MM (DFT/MM) | B3LYP/cc-pVDZ, CHARMM | ~100 - 200 mV | Accurate treatment of hydrogen bonding & charge transfer |
Experimental Protocols for Validation
Protocol for Experimental pKa Determination via Potentiometric Titration
Principle: Measurement of solution pH as a function of added acid or base to determine proton dissociation constants. Materials: Analyte compound, high-purity water or mixed solvent, standardized HCl and KOH solutions, pH meter with glass electrode, thermostatted titration vessel, inert atmosphere (N₂/Ar) supply. Procedure:
- Prepare a 1-5 mM solution of the analyte in 0.1 M KCl (for constant ionic strength). De-gas with inert gas.
- Calibrate the pH meter at the experimental temperature using standard buffers (pH 4.01, 7.00, 10.01).
- Initial pH Measurement: Record the starting pH of the analyte solution.
- Titration: Using an automated titrator or burette, add small increments (e.g., 0.05 mL) of standardized KOH (for acidic compounds) or HCl (for basic compounds).
- Equilibrium: After each addition, stir and wait until a stable pH reading is obtained (signal drift <0.01 pH/min).
- Data Processing: Fit the pH vs. volume data (titration curve) using nonlinear regression software (e.g., Hyperquad, BEST7) to solve the equilibrium equations and extract pKa values. For multiprotic systems, global fitting is required.
Protocol for Experimental Redox Potential Determination via Cyclic Voltammetry (CV)
Principle: Application of a linear voltage sweep to an electrochemical cell and measurement of the resulting current to identify reduction and oxidation peaks. Materials: Analyte, supporting electrolyte (e.g., 0.1 M TBAPF6 in acetonitrile or buffer), working electrode (glassy carbon, Pt), reference electrode (Ag/AgCl or SCE), counter electrode (Pt wire), potentiostat, electrochemical cell, inert atmosphere (N₂/Ar) supply. Procedure:
- Cell Preparation: Clean the working electrode thoroughly (e.g., polish with 0.05 μm alumina slurry). Add supporting electrolyte solution to the electrochemical cell.
- Deoxygenation: Sparge the solution with inert gas for at least 15 minutes to remove dissolved oxygen. Maintain a blanket of gas during the experiment.
- Reference Calibration: Record the CV of a standard redox couple (e.g., ferrocene/ferrocenium, Fc/Fc⁺) under identical conditions to calibrate the reference electrode potential.
- Sample Measurement: Add the analyte to the cell. Apply a linear potential sweep between designated start and end potentials at a defined scan rate (e.g., 100 mV/s).
- Data Analysis: Identify the cathodic (reduction) and anodic (oxidation) peak potentials (Epc and Epa). The formal redox potential E⁰' is approximated as (Epa + Epc)/2. Verify reversibility by checking the peak separation (ΔE_p ≈ 59 mV for a reversible one-electron process at 25°C) and scan rate independence.
Visualization of Workflows and Relationships
Workflow for Computational pKa Prediction and Validation
Key Challenges in Biomolecular Redox Prediction
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for Experimental Validation
| Item | Function/Application | Key Considerations |
|---|---|---|
| TRIS Buffer | Biological pH buffering (pH 7-9) for pKa titrations & redox protein studies. | Metal-free, high-purity grade to avoid redox-active contaminants. |
| Potassium Chloride (KCl) | Provides constant ionic strength in potentiometric titrations. | Analytical grade, dried to control water content. |
| Tetrabutylammonium Hexafluorophosphate (TBAPF6) | Supporting electrolyte for non-aqueous electrochemical studies (CV). | Must be rigorously purified (recrystallized) to remove electroactive impurities. |
| Ferrocene (Fc) | Internal reference standard for redox potential calibration in non-aqueous CV. | Redox potential is solvent-dependent; report vs. Fc/Fc⁺. |
| Dithiothreitol (DTT) / Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agents to maintain protein thiols in reduced state for redox studies. | TCEP is more stable and metal-chelator compatible. |
| Deuterated Buffers (e.g., D₂O pD buffers) | For NMR-based pKa determination, minimizing solvent proton interference. | pD = pH meter reading + 0.4. |
| Anaerobic Chamber Glove Box | Creates oxygen-free environment for handling air-sensitive compounds in redox studies. | Maintains <1 ppm O₂; essential for studying low-potential species. |
| Quartz Electrochemical Cell | For UV-Vis spectroelectrochemistry, allowing simultaneous spectral and potential control. | Must be compatible with both potentiostat and spectrometer cuvette holders. |
The Hohenberg-Kohn (HK) theorems established the theoretical bedrock for modern density functional theory (DFT). The first theorem proves that the ground-state electron density uniquely determines all properties of a many-electron system, while the second provides a variational principle for determining the exact ground-state density. This framework shifted the paradigm from wavefunction-based methods to density-based computations, enabling practical electronic structure calculations for real materials. However, the accuracy of DFT is fundamentally limited by the approximations inherent in the exchange-correlation functional. This whitepaper posits that the logical evolution of this foundational work is the integration of DFT with machine learning (ML) to create high-fidelity, computationally scalable potentials. Here, DFT's role transitions from a direct computational tool to a generator of high-quality training data and a physical scaffold for machine-learned potentials (MLPs), merging first-principles rigor with data-driven efficiency.
Theoretical Foundation: From Hohenberg-Kohn to Hybrid Potentials
The HK theorems provide the necessary legitimacy for using electron density as the central variable. In the context of MLPs, this implies that an accurate representation of the atomic environment, which indirectly encodes the electron density, should be sufficient to predict total energies and forces. The hybrid approach uses DFT to calculate the exact (within its functional approximation) energy, forces, and stress tensors for a carefully chosen set of atomic configurations. This dataset then trains an ML model—typically a neural network or Gaussian approximation potential—to interpolate and extrapolate this potential energy surface (PES) with near-DFT accuracy but at a fraction of the computational cost for molecular dynamics (MD) and sampling.
Core Methodologies and Protocols
Protocol for Generating the Foundational DFT Dataset
A robust MLP requires a comprehensive, diverse, and thermodynamically consistent training set.
System Sampling:
- Perform ab initio MD (AIMD) using DFT (e.g., VASP, Quantum ESPRESSO) at various thermodynamic conditions (temperatures, pressures) to sample relevant phases and metastable states.
- Use enhanced sampling techniques (e.g., metadynamics, umbrella sampling) to probe rare events and transition states.
- Generate random cell distortions and atomic displacements to include off-equilibrium configurations.
- Include surfaces, vacancies, and defect structures relevant to the application.
DFT Calculation Parameters:
- Functional: Select based on system (e.g., PBEsol for solids, SCAN for accuracy, PBE-D3 for dispersion).
- Basis Set/Plane-wave Cutoff: Ensure convergence of total energy (< 1 meV/atom).
- k-point Grid: Use a Monkhorst-Pack grid dense enough for energy convergence.
- Convergence Criteria: Energy: 10^−6 eV; Force: 0.01 eV/Å.
Data Curation: Remove duplicate configurations using structure matching (e.g., SOAP similarity). The final dataset should contain 10^3 to 10^5 configurations, each with lattice vectors, atomic positions, species, total energy, atomic forces, and virial stresses.
Protocol for Training a Machine-Learned Potential
Descriptor Generation: Convert atomic configurations into invariant descriptors. Common choices are:
- Atomic Cluster Expansion (ACE)
- Smooth Overlap of Atomic Positions (SOAP)
- Behler-Parrinello Symmetry Functions
Model Architecture & Training:
- Architecture: Use a deep neural network (e.g., 3 layers, 64 nodes/layer) or a moment tensor potential (MTP) framework.
- Loss Function: L = w_E * MSE(E) + w_F * MSE(F) + w_S * MSE(S), where E, F, S are energy, force, and stress, respectively. Weights (w) are tuned to balance the magnitude of each component.
- Training: Split data 80:10:10 (training:validation:test). Use Adam optimizer with early stopping based on validation loss.
Active Learning Protocol: To mitigate poor extrapolation, an active learning loop is essential.
- Run MD with the provisional MLP.
- Detect "uncertain" configurations where the MLP's ensemble predictions have high variance.
- Compute DFT single-point calculations for these uncertain configurations.
- Augment the training set and retrain the model. Iterate until no uncertain configurations are found under operational conditions.
Table 1: Comparison of Computational Cost and Accuracy for Si System (256 atoms)
| Method | Computational Cost (CPU-hrs / 1 ps MD) | Force Error (meV/Å) (MAE) | Energy Error (meV/atom) (MAE) | Typical Use Case |
|---|---|---|---|---|
| DFT (PBE) | 2,000 - 5,000 | 0 (Reference) | 0 (Reference) | Small-scale, high-accuracy validation |
| Classical Force Field (SW) | < 0.1 | 50 - 200 | 5 - 20 | Large-scale, low-accuracy screening |
| MLP (trained on DFT) | 1 - 10 | 10 - 30 | 1 - 5 | Mesoscale dynamics with near-DFT fidelity |
Table 2: Performance of Representative MLP Frameworks (2023-2024 Benchmark Data)
| Framework | Descriptor Type | Key Strengths | Best-Suported Systems |
|---|---|---|---|
| DeePMD-kit | Deep Neural Network | High performance, scalable to >100M atoms | Bulk materials, molecules, interfaces |
| MACE | Atomic Cluster Expansion | High body-order accuracy | Complex molecules, reactive chemistry |
| GAP / QUIP | Smooth Overlap (SOAP) | Strong mathematical foundation, interpretability | Amorphous materials, allotropes |
| ANI (Neurochem) | Modified AEV (Atomic Env.) | Optimized for organic molecules & drug-like systems | Biochemical molecules, drug candidates |
Visualizations
(Title: Hybrid DFT-ML Potential Development Workflow)
(Title: Logical Bridge from HK Theorems to ML Potentials)
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Solution | Function in Hybrid DFT-MLP Workflow | Example / Note |
|---|---|---|
| DFT Software Suite | Generates the foundational training data (energies, forces). | VASP, Quantum ESPRESSO, CP2K, ABINIT. Choice depends on system type and functional needs. |
| ML Potential Framework | Provides the architecture and code to train and deploy the MLP. | DeePMD-kit, MACE, AMPtorch (PyTorch), QUIP/GAP. Open-source with active communities. |
| Atomic Descriptor Library | Translates atomic coordinates into rotationally & permutationally invariant inputs for the ML model. | DScribe, quippy (for SOAP), internal functions within MLP frameworks. |
| Ab Initio MD Engine | Samples the potential energy surface for initial data generation and active learning. | LAMMPS (with DFTB/ML plug-ins), i-PI, ASE Molecular Dynamics module. |
| Active Learning Manager | Automates the uncertainty quantification and iterative training loop. | FLARE, ALKEMIE, custom scripts using model ensembles or Bayesian uncertainty. |
| High-Performance Computing (HPC) Cluster | Essential for both DFT data generation (high cost) and large-scale MLP-MD production runs. | CPU nodes for DFT, GPU nodes (NVIDIA A100/H100) for efficient MLP training and inference. |
| Reference Database | Pre-computed, high-quality DFT datasets for common systems to bootstrap training. | Materials Project, NOMAD, OC20, QM9. Reduces initial computational overhead. |
The development of accurate, computationally tractable density functional theory (DFT) methods remains a central challenge in quantum chemistry and materials science for simulating complex systems like molecular crystals, proteins, and catalytic surfaces. This pursuit is fundamentally grounded in the Hohenberg-Kohn (HK) theorems, which establish the electron density as the basic variable and guarantee the existence of a universal functional for the energy. However, the exact form of the exchange-correlation (XC) functional is unknown, leading to the "Jacob's Ladder" of approximations. This whitepaper focuses on the highest, most sophisticated rungs: Range-Separated Hybrids (RSHs) and Non-Empirical Functionals, which are designed to overcome critical failures of standard DFT—particularly delocalization error, self-interaction error, and poor description of non-covalent interactions—without resorting to empirical parameter fitting.
Core Concepts: Range-Separated Hybrids and Non-Empirical Design
Range-Separated Hybrid Functionals
RSHs partition the electron-electron interaction into short-range (SR) and long-range (LR) components using a smooth function, typically the error function complemented by a parameter, ω (the range-separation parameter).
[ \frac{1}{r{12}} = \frac{1 - \text{erf}(\omega r{12})}{r{12}} + \frac{\text{erf}(\omega r{12})}{r_{12}} ]
The first term is SR, the second is LR. The key innovation is the use of different approximations for each range:
- Short-Range: Treated with a (semi-)local DFT functional to capture dynamic correlation.
- Long-Range: Treated with exact Hartree-Fock (HF) exchange to correct for self-interaction and improve the description of charge-transfer and Rydberg states.
Popular RSHs include ωB97X-V, LC-ωPBE, and CAM-B3LYP. The "optimal" value of ω can be determined non-empirically, e.g., by enforcing the ionization Potential theorem.
Non-Empirical Functional Construction
Non-empirical ("first-principles" or "ab initio") functionals are constructed to satisfy as many exact theoretical constraints as possible (e.g., the uniform electron gas limit, coordinate scaling relations, the IP theorem) without fitting parameters to experimental or high-level ab initio databases. The SCAN (Strongly Constrained and Appropriately Normed) meta-GGA functional is a landmark, satisfying 17 known exact constraints. Its successor, rSCAN, improves numerical stability. These functionals can serve as the base for RSHs (e.g., ωB97M-V) or double-hybrids, creating a powerful, parameter-free hierarchy.
Quantitative Performance and Benchmark Data
The performance of leading functionals is assessed against benchmark databases like GMTKN55 (general main-group thermochemistry, kinetics, and noncovalent interactions), S22 (non-covalent complexes), and TMC34 (transition metal complexes). Recent searches (2024) confirm the superior accuracy of modern RSHs and non-empirical functionals.
Table 1: Performance of Selected State-of-the-Art Functionals on Key Benchmark Sets (Mean Absolute Deviation, MAD)
| Functional Class | Example Functional | GMTKN55 MAD [kcal/mol] | S22 MAD [kcal/mol] | TMC34 MAD [kcal/mol] | Key Strength |
|---|---|---|---|---|---|
| Non-Empirical Meta-GGA | SCAN | 5.2 | 0.5 | 6.8 | Solid-state, constraint satisfaction |
| Non-Empirical Hybrid | SCAN0 | 4.1 | 0.4 | 5.9 | Improved band gaps, surfaces |
| Range-Separated Hybrid | ωB97M-V | 3.6 | 0.2 | 5.2 | Excellent across diverse chemistries |
| Double Hybrid (RSH) | ωB97M(2) | 3.3 | 0.2 | 4.8 | Top-tier accuracy, higher cost |
Data synthesized from recent literature and benchmark repositories (2023-2024). Lower MAD indicates better performance.
Table 2: Comparison of Range-Separation Parameter Determination Methods
| Method | Description | Example Functional | Key Advantage |
|---|---|---|---|
| System-Dependent Tuning | ω tuned per system to satisfy IP theorem via ΔSCF. | OT-ωB97X | Eliminates delocalization error for specific system (e.g., dyes). |
| Non-Empirical Optimization | ω optimized globally to satisfy multiple constraints. | ωB97X-V | Robust, general-purpose; no need for system-specific tuning. |
| Damped Response | Uses a universal damping parameter for LR. | DSD-PBEP86 | Improves accuracy for non-covalent interactions. |
Experimental and Computational Protocols
Protocol for Non-Empirical Functional Assessment (GMTKN55 Benchmark)
- System Preparation: Obtain geometries for all 55 subsets (~1500 calculations) from the GMTKN55 database.
- Computational Setup: Use a robust quantum chemistry code (e.g., ORCA, Q-Chem, Gaussian). Employ a large, diffuse basis set (e.g., def2-QZVP) with matching auxiliary basis for RI/JK acceleration.
- Energy Calculations: Perform single-point energy calculations on all systems using the target functional (e.g., ωB97M-V) and a reference high-level method (e.g., DLPNO-CCSD(T)).
- Statistical Analysis: Compute relative energies for each reaction subset. Calculate the Mean Absolute Deviation (MAD) and Root-Mean-Square Deviation (RMSD) for each subset and the overall weighted total (WTMAD-2).
Protocol for System-Specific Optimal Tuning (OT-RSH)
- Initial Guess: Select an initial ω value (e.g., 0.2 Bohr⁻¹) and a basis set suitable for anions (e.g., aug-cc-pVTZ).
- ΔSCF Calculations: Calculate the ionization potential (IP) as E(N-1) - E(N) and the electron affinity (EA) as E(N) - E(N+1) for the system of interest (e.g., an organic semiconductor molecule).
- Condition Enforcement: Iteratively adjust ω until the condition -εHOMO(N) ≈ IP is satisfied (where εHOMO is the highest occupied molecular orbital energy). The condition -ε_LUMO(N) ≈ EA is often monitored concurrently.
- Validation: Use the tuned functional to calculate the target property (e.g., charge-transfer excitation energy).
Visualizing the Functional Development and Assessment Workflow
Diagram 1: DFT Functional Development Hierarchy
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Advanced DFT Research
| Item/Category | Example(s) | Function/Purpose |
|---|---|---|
| Quantum Chemistry Software | ORCA, Q-Chem, Gaussian, PySCF, NWChem | Primary engines for performing DFT and wavefunction calculations with support for advanced functionals. |
| Basis Set Libraries | def2 series (def2-TZVP, def2-QZVP), aug-cc-pVXZ, jun-cc-pVXZ | Sets of mathematical functions describing electron orbitals; critical for accuracy, especially for non-covalent interactions. |
| Benchmark Databases | GMTKN55, S22, S66, TMC34, Iowa Database | Curated sets of molecular systems and reference data for validating and benchmarking new functionals. |
| Analysis & Visualization | Multiwfn, VMD, Jmol, ChemCraft | Software for analyzing electron density, orbitals, non-covalent interactions (NCI plots), and visualizing molecular structures. |
| Automation & Scripting | Python (with NumPy, SciPy), Bash, ASE (Atomistic Simulation Environment) | For automating workflows (e.g., parameter tuning, batch calculations) and data processing. |
| High-Performance Computing (HPC) | Local clusters, Cloud computing (AWS, GCP), National supercomputing centers | Provides the necessary computational power for large-scale simulations of complex systems. |
Conclusion
The Hohenberg-Kohn theorems provide the rigorous, elegant foundation that makes Density Functional Theory the workhorse of computational quantum chemistry and materials physics. By establishing electron density as the fundamental variable, they enabled the scalable simulations essential for modern research, from screening drug candidates to designing novel catalysts. While challenges remain—particularly in systematically improving exchange-correlation approximations and capturing delicate electronic correlations—the framework is remarkably adaptable. The future lies in its integration with emerging data-driven techniques. For biomedical and clinical researchers, this evolution promises more accurate predictions of in-vitro and in-vivo behavior, accelerating rational drug design and the development of advanced biomaterials by providing unprecedented atomic-level insight into molecular structure, interaction, and function.